SECURITIES
AND EXCHANGE COMMISSION
WASHINGTON,
D.C. 20549
FORM
S-1
REGISTRATION
STATEMENT UNDER
THE
SECURITIES ACT OF 1933
ANEW
MEDICAL, INC.
(Exact
name of registrant as specified in its charter)
| Delaware | | 2836 | | 86-2727441 |
(State or other jurisdiction of
incorporation or organization) | | (Primary Standard Industrial
Classification Code) | | (I.R.S. Employer
Identification No.) |
13576
Walnut Street, Suite A
Omaha, NE 68144
Telephone:
(833) 931-6330
(Address
and telephone number of registrant’s
principal
executive offices)
Joseph
A. Sinkule
13576
Walnut Street, Suite A
Omaha, NE 68144
Telephone:
(833) 931-6330
(Name,
address, including zip code, and telephone number,
including
area code, of agent for service)
Copies
of all Correspondence to:
Paul
Goodman, Esq.
Cyruli
Shanks & Zizmor, LLP
Attorneys
At Law
420
Lexington Avenue
Suite
2320
New
York, NY 10170
Telephone:
(212) 212-6800
Approximate
date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If
any of the securities being registered on this form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the
Securities Act of 1933, please check the following box: ☒
If
this form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following
box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.
☐
If
this form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If
this form is a post-effective amendment filed pursuant to Rule 462 (d) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If
delivery of the prospectus is expected to be made pursuant to Rule 434, please check the following box: ☐
Indicate by check mark whether the registrant is a large accelerated
filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions
of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging
growth company” in Rule 12b-2 of the Exchange Act.
| | Large accelerated filer | ☐ | | Accelerated filer | ☐ |
| | Non-accelerated filer | ☒ | | Smaller reporting registrant | ☒ |
| | | | Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
(1)
We are registering (a) up to 11,500,000 shares of common stock that we may issue to investors upon the exercise of warrants originally
issued as part of a unit sold in our Initial Public Offering, and (b) 10,027,925 of common stock currently held by certain other selling
shareholders which includes (i) up to 415,000 shares of common stock that we may issue to Redwoods, LLC upon the exercise of warrants
and (ii) up to 115,000 shares of common stock that we may issue to Chardan Capital Markets, LLC upon the exercise of warrants.
(2)
The offering price has been estimated solely for the purpose of computing the amount of the registration fee in accordance with Rule
457(c) of the Securities Act, based upon the average of the high and low sales prices for the registrant’s common stock as reported
by Nasdaq on August 30, 2024.
The
registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the
registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective
in accordance with Section 8(a) of the Securities Act of 1933 or until this registration statement shall become effective on such date
as the Commission, acting pursuant to said Section 8(a), may determine.
THE
INFORMATION IN THIS PROSPECTUS IS NOT COMPLETE AND IS SUBJECT TO COMPLETION AND MAY BE CHANGED. WE MAY NOT SELL THESE SECURITIES UNTIL
THE REGISTRATION STATEMENT FILED WITH THE SECURITIES AND EXCHANGE COMMISSION IS EFFECTIVE. THIS PROSPECTUS IS NOT AN OFFER TO SELL THESE
SECURITIES AND IT IS NOT SOLICITING AN OFFER TO BUY THESE SECURITIES IN ANY STATE WHERE THE OFFER OR SALE IS NOT PERMITTED.
Preliminary
Prospectus Dated September 5, 2024
ANEW
MEDICAL, INC.
Up
to a Maximum of 21,527,925 Shares of Common Stock
This prospectus relates to the offering and resale
by the selling stockholders identified herein of up to 21,527,925 shares of common stock, par value $0.0001 per share, of ANEW Medical
Inc. These shares include (a) up to 11,500,000 shares of common stock that we may issue to investors upon the exercise of warrants originally
issued as part of a unit sold in our Initial Public Offering, and (b) 10,027,925 of common stock currently held by certain other selling
shareholders which includes (i) up to 415,000 shares of common stock that we may issue to Redwoods, LLC upon the exercise of warrants
and (ii) up to 115,000 shares of common stock that we may issue to Chardan Capital Markets, LLC upon the exercise of warrants.
The Selling Stockholders may offer all or part
of the shares for resale from time to time through public or private transactions at either fixed prices or prevailing market prices at
the time of sale, at varying prices or negotiated prices.
Any
broker-dealers or agents that are involved in such resales may be deemed to be “underwriters” within the meaning of the Securities
Act in connection therewith. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of
the shares purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. For more information,
please see the section of this prospectus titled “Plan of Distribution” beginning on page 30.
We
will not receive any proceeds from the resale of shares of common stock by the Selling Stockholders.
Our
common stock is traded on the NASDAQ Global Market under the symbol “WENA.” On August 30, 2024, the average of the high and
low sales prices of our common shares was $0.95 per share. These prices will fluctuate based on the demand for our common shares.
Investing
in our common shares involves a high degree of risk. You should purchase shares only if you can afford a complete loss. See “Risk
Factors” beginning on page 2.
Neither
the Securities and Exchange Commission (“SEC”) nor any state securities commission has approved or disapproved of these securities
or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
TABLE
OF CONTENTS
Unless
otherwise specified or the context otherwise requires, references in this prospectus to “we”, “our” and “us”
and the “Company” refer to ANEW Medical, Inc.
PROSPECTUS
SUMMARY
To
understand this offering fully, you should read the entire prospectus carefully, including the risk factors beginning on page 2 and the
consolidated financial statements.
This
summary highlights selected information appearing elsewhere in this prospectus. While this summary highlights what we consider to be
important information about us, you should carefully read this entire prospectus before investing in our Common Stock, especially the
risks and other information we discuss under the headings “Risk Factors” and “Management’s Discussion and Analysis
of Financial Condition and Results of Operations” and our consolidated financial statements and related notes beginning on page
60. Our fiscal year end is December 31 and our fiscal years ended December 31, 2023 and 2022 are sometimes referred to herein as fiscal
years 2023 and 2022, respectively. Some of the statements made in this prospectus discuss future events and developments, including our
future strategy and our ability to generate revenue, income and cash flow. These forward-looking statements involve risks and uncertainties
which could cause actual results to differ materially from those contemplated in these forward-looking statements. See “Forward-Looking
Statements”. Unless otherwise indicated or the context requires otherwise, the words “we,” “us,” “our”,
the “Company” or “our Company” or “ANEW” refer to ANEW Medical, Inc, a Delaware corporation, and
our each of our subsidiaries.
| Issuer: |
|
ANEW
Medical, Inc., a Delaware corporation. |
| |
|
|
Shares
of Common Stock
offered
by the Selling Stockholders (1): |
|
10,027,925 |
| |
|
|
Shares
of Common Stock
Outstanding
Prior to the Offering (1) |
|
21,263,515 |
| |
|
|
Terms
of Offering
|
|
The
Selling Stockholders will determine when and how they will sell shares of common stock offered
in this prospectus. |
| |
|
|
| Use
of Proceeds |
|
We
will not receive any proceeds from the sale of the common stock by the Selling Stockholders.
All net proceeds from the sale of the common shares covered by this prospectus will go to
the Selling Stockholders. However, we may receive the proceeds from the exercise of any warrants
if the Selling Stockholders do not exercise the Warrants on a cashless basis, if and when
exercised. See the section of this prospectus titled “Use of Proceeds.” |
| |
|
|
| Trading
Symbol |
|
Our
shares of common stock are listed on The Nasdaq Global Market under the symbol “WENA” |
| |
|
|
| Transfer
Agent |
|
Continental
Stock Transfer and Trust Company |
| |
|
|
| Dividend
Policy |
|
We
have never declared or paid any cash dividends on our shares of common stock. We do not anticipate
paying any cash dividends on our common stock in the foreseeable future. |
| |
|
|
| Risk
Factors |
|
The
shares of common stock offered hereby involve a high degree of risk and should not be purchased
by investors who cannot afford the loss of their entire investment. |
| (1) | As
of August 30, 2024. Common Stock offered by the Selling Stockholders excludes 11,500,000
shares of our Common Stock issuable upon the exercise of the Public Warrants. |
RISK
FACTORS
This
investment has a high degree of risk. Before you invest you should carefully consider the risks and uncertainties described below and
the other information in this prospectus. If any of the following risks actually occur, our business, operating results and financial
condition could be harmed and the value of our stock could go down. This means you could lose all or a part of your investment. You
should carefully consider the risks described below together with all of the other information included in our public filings before
making an investment decision with regard to our securities. The statements contained in or incorporated into this document that are
not historic facts are forward-looking statements that are subject to risks and uncertainties that could cause actual results to differ
materially from those set forth in or implied by forward-looking statements. If any of the following events described in these risk factors
actually occur, our business, financial condition or results of operations could be harmed. In that case, the trading price of our common
stock could decline, and you may lose all or part of your investment. Moreover, additional risks not presently known to us or that we
currently deem less significant also may impact our business, financial condition or results of operations, perhaps materially. For additional
information regarding risk factors, see “Forward-Looking Statements”.
Risks
Related to Our Business and Operations
Our
business is subject to a number of risks of which you should be aware before making decision to invest in our common stock
These
risks are more fully described in this “Risk Factors” section, including the following:
| ● | We
have incurred significant losses since our inception. We expect to incur losses over the
next several years and may never achieve or maintain profitability. |
| ● | We
will need substantial additional funding to meet our financial obligations and to pursue
our business objectives. |
| ● | We
currently primarily rely on our collaborations with Reliance Life Sciences Ltd. (“RLS”)
and Universitat Autònoma de Barcelona (“UAB”), for our preclinical research
and development programs. |
| ● | Negative
public opinion of gene therapy and increased regulatory scrutiny of gene therapy and genetic
research may adversely impact the development or commercial success of our current and future
product candidates. |
| ● | We
face significant competition in the area of biosimilar biologics from other biotechnology
and pharmaceutical companies. |
| ● | We
use AAV and lentiviral vectors, and lipid nanoparticles to delivery some gene therapies,
but others may have property rights to these vectors that we may need to license. |
We
are dependent on the success of our biosimilar and gene therapy product candidates, which are still in early stages of development.
We
currently have no products that are approved for commercial sale and may never be able to develop marketable products. We expect that
a substantial portion of our efforts and expenditures over the next few years will be devoted to the development of our biosimilar
product candidates, which are in the early stages of clinical development, and the gene therapy product candidates, all of which are
in pre-clinical development. Accordingly, our business currently depends heavily on the successful development, regulatory approval and
commercialization of these product candidates. We cannot be certain that any of our product candidates will receive regulatory approval
or be successfully commercialized even if we receive regulatory approval.
We
will require additional capital to fund our operations, and if we fail to obtain necessary financing, we may not be able to complete
the development and commercialization of our product candidates.
We
expect to continue to incur significant operating and net losses, as well as negative cash flows from operations, for the foreseeable
future as we continue to develop our gene therapy product candidates and prepare for potential future regulatory approvals and commercialization
of our products. We have not generated any revenue to date and does not expect to generate product revenue unless and until we successfully
complete development and obtain regulatory approval for at least one of our gene therapy product candidates. Our current cash and cash
equivalents balance will also not be sufficient to complete all necessary development activities and commercially launch our products.
We
are unable to estimate the actual funds that we will require for development and any approved marketing and commercialization activities.
We
expect to spend substantial amounts to complete the development of, seek regulatory approvals for and commercialize our product candidates.
Because the length of time and activities associated with successful development of our product candidates is highly uncertain, we are
unable to estimate the actual funds we will require for development and any approved marketing and commercialization activities. Our
future funding requirements, both near and long- term, will depend on many factors, including, but not limited to:
| ● | the
progress, timing, costs and results of our clinical trials of our product candidates; |
| ● | the
outcome, timing and cost of meeting regulatory requirements; |
| ● | the
achievement of certain development, regulatory and commercialization milestones |
| ● | the
cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual
property rights; |
| ● | the
cost of obtaining necessary intellectual property and defending potential intellectual property
disputes s brought by third parties against us; |
| ● | the
effect of competing technological and market developments; |
| ● | the
cost and timing of completion of clinical-stage and commercial-scale manufacturing activities; |
| ● | the
cost of establishing sales, marketing and distribution capabilities for our product candidates;
and |
| ● | the
initiation, progress, timing and results of our commercialization of our product candidates. |
We
may be required to make significant payments to third parties under the agreements pursuant to which e acquired our gene therapy and
biosimilar product candidates.
Under
our agreements with RLS and UAB, we are subject to significant obligations, including payment obligations upon achievement of specified
milestones and payments based on product sales, as well as other material obligations. Some of these milestones require us to make payments
prior to generating any product sales. We may not have sufficient funds available to meet our obligations at such time as any of these
payments become due, in which case our development efforts would be harmed. Further, failure to make these payments or to meet our other
material obligations may result in our counterparties pursuing various remedies under those agreements that could harm our operations.
We
are highly dependent on the services of our key executives and personnel, including our Chief Executive Officer, Dr. Joseph Sinkule
(Pharm. D.) and if we are not able to retain these members of our management or recruit additional management, clinical and scientific
personnel, our business will suffer.
We
are highly dependent on the principal members of our management and scientific and technical staff, particularly, our Chief Executive
Officer, Dr. Joseph Sinkule. Dr. Sinkule’s current employment agreement with us expires in 2024. The loss of service
of any of our management or key scientific and business management staff could harm our business. In addition, we are dependent on our
continued ability to attract, retain and motivate highly qualified additional management, clinical and scientific personnel. If we are
not able to retain our management and to attract, on acceptable terms, additional qualified personnel necessary for the continued development
of our business, we may not be able to sustain our operations or grow.
We
need to attract and retain key personnel.
We
may not be able to attract or retain qualified management and commercial, scientific and clinical personnel due to the intense competition
for qualified personnel among biotechnology, pharmaceutical and other businesses, and the cost of living. If we are not able to attract
and retain necessary personnel to accomplish our business objectives, we may experience constraints that will impede the achievement
of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
Our
senior management and key employees may terminate their position with us at any time. If we lose one or more members of our senior management
team or key employees, our ability to successfully implement our business strategy could be seriously harmed. Replacing these individuals
may be difficult, cause disruption, and may take an extended period of time because of the limited number of individuals in our industry
with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition
to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate additional key personnel. We does
not maintain “key person” insurance for any of our executives or other employees.
Many
of the other biopharmaceutical companies that we compete against for qualified personnel and consultants have greater financial and other
resources, different risk profiles and a longer history in the industry than we do. They also may provide more diverse opportunities
and better chances for career advancement. Some of these characteristics may be more appealing to high- quality candidates and consultants
than what we have to offer. If we are unable to continue to attract and retain high-quality personnel and consultants, the rate and success
at which we can discover and develop product candidates, our ability to effectively manage any future growth and our business will be
limited.
Our
employees, independent contractors, principal investigators, consultants, commercial collaborators, manufacturers, service providers
and other vendors, or those of our affiliates, may engage in misconduct or other improper activities.
Misconduct
by any of these parties could include intentional, reckless and/or negligent conduct or other unauthorized activities that violate the
laws and regulations, including those of the FDA and other similar regulatory bodies that require the reporting of true, complete and
accurate information; manufacturing standards; federal, state and foreign healthcare fraud and abuse laws and data privacy; or laws that
require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and other business
arrangements in the healthcare industry are subject to extensive laws intended to prevent fraud, kickbacks, self-dealing, bribery, corruption,
antitrust violations, and other abusive practices. These laws may restrict or prohibit a wide range of business activities, including
research, manufacturing, distribution, pricing, discounting, marketing and promotion, sales commission, customer incentive programs and
other business arrangements. It is not always possible to identify and deter employee or third-party misconduct, and the precautions
we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting
our from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations.
Additionally,
we are subject to the risk that a person or government agency could allege such fraud or other misconduct, even if none occurred. If
any of our employees, independent contractors, principal investigators, consultants, commercial collaborators, manufacturers, service
providers or other vendors, or those of our affiliates, are alleged or found to be in violation of any such regulatory standards or requirements,
or become subject to a corporate integrity agreement or similar agreement and curtailment of our operations, it could have a significant
impact on our business and financial results, including the imposition of significant civil, criminal and administrative penalties, damages,
monetary fines, suspension or delay in clinical trials, possible exclusion from participation in Medicare, Medicaid and other federal
healthcare programs, FDA debarment, contractual damages, reputational harm, diminished profits and future earnings, additional reporting
requirements and oversight, any of which could adversely affect our ability to operate our business and our results of operations.
Operation
of our business internationally exposes us to business, regulatory, political, operational, financial and economic risks associated with
doing business in various jurisdictions globally.
Our
business strategy includes establishing and maintaining operations and certain key third party arrangements in various jurisdictions
around the world. Doing business internationally involves a number of risks, including:
| ● | multiple,
conflicting and changing laws and regulations such as tax laws, export and import restrictions,
employment laws, anti-bribery and anti- corruption laws, regulatory requirements and other
governmental approvals, permits and licenses; |
| ● | failure
by us or our distributors to obtain appropriate licenses or regulatory approvals for the
sale or use of our product candidates, if approved, in various countries; |
| ● | difficulties
in managing foreign operations; |
| ● | unexpected
changes in tariffs or trade barriers; |
| ● | complexities
associated with managing multiple payor-reimbursement regimes or self-pay systems; |
| ● | financial
risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts
receivable and exposure to foreign currency exchange rate fluctuations; |
| ● | reduced
protection for intellectual property rights; |
| ● | reduced
protection of contractual rights in the event of bankruptcy or insolvency of the other contracting
party; |
| ● | natural
disasters, political and economic instability, including wars, terrorism and political unrest,
outbreak of disease, including the recent COVID-19 pandemic and related shelter-in-place
orders, travel, social distancing and quarantine policies, boycotts, curtailment of trade
and other business restrictions; |
| ● | failure
to comply with foreign laws, regulations, standards and regulatory guidance governing the
collection, use, disclosure, retention, security and transfer of personal data, including
the European Union General Data Privacy Regulation (“GDPR”); and |
| ● | failure
to comply with anti-bribery and anti-corruption laws including the Foreign Corrupt Practices
Act, including its books and records provisions and its anti-bribery provisions, including
by failing to maintain accurate information and control over sales and distributors’
activities. |
Any
of these risks, if encountered, could significantly harm our current or future international operations and, consequently, negatively
impact our financial condition, results of operations and cash flows.
Risks
Related to Development of Biosimilar Biologics and Cell and Gene Therapies
If
we are unable to obtain regulatory approval for, or successfully commercialize, bevacizumab-anew, or rituximab-anew, our business will
be harmed.
Biosimilar
product development is a highly speculative undertaking and involves a substantial degree of risk. We have initiated preparatory activities
for our confirmatory Phase 3 clinical trial of bevacizumab-anew (Avastin) biosimilar candidate, and rituximab-anew (Rituxan/Mabthera)
candidate. It may be several years, if ever, before we complete Phase 3 clinical trials and has a product candidate ready to
file for market approval with the relevant regulatory agencies. If we obtain regulatory approval to market a biosimilar product candidate,
our future revenue will depend upon the size of any markets in which our product candidates may receive approval and our ability to achieve
sufficient market acceptance, pricing, reimbursement from third-party payors and adequate market share for our product candidates in
those markets. Even if one or more of our product candidates gain regulatory approval and are commercialized, we may never become profitable.
Our biosimilar product candidates are for versions of products for which other approved biosimilar competitors are already being marketed.
Thus, even if approved, our products will face significant established competition from both the original branded version of our products,
as well as from other already-approved biosimilar products. We cannot be certain that any of our product candidates will be successful
in clinical trials or receive regulatory approval. Further, our product candidates may not receive regulatory approval even if they are
successful in clinical trials. If we do not receive regulatory approvals for our product candidates, we may not be able to continue our
operations.
The
evolving regulatory approval processes of the FDA, EMA and comparable foreign authorities are lengthy, time-consuming, rigorous and inherently
unpredictable. If we and our collaboration partners are ultimately unable to obtain regulatory approval for our product candidates, our
business will be harmed.
The
research, development, testing, manufacturing, labeling, packaging, approval, promotion, advertising, storage, marketing, distribution,
post-approval monitoring and reporting and export and import of biologic products are subject to extensive regulation by the FDA and
other regulatory authorities in the United States, by the EMA and Competent Authorities in the EEA, and by other regulatory authorities
in other countries, where regulations differ from country to country. We are not permitted to market our product candidates in the United States
until we receive approval from the FDA, or in the EEA until we receive European Commission or EEA Competent Authority approvals.
The
exact amount of time required to obtain approval by the FDA and comparable foreign authorities is unpredictable, may take years
and depends upon numerous factors, which may not be within our control. In addition, approval policies, regulations or the type and amount
of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may
vary among jurisdictions, which could cause delays in the approval or the decision not to approve an application. We have not obtained
regulatory approval for any of our product candidates, and it is possible that none of our current or future product candidates will
ever obtain regulatory approval.
This
lengthy approval process, as well as the unpredictability of the results of clinical trials, may result in our failure to obtain regulatory
approval to market any of our product candidates, which would significantly harm our business. Moreover, any delays in the commencement
or completion of clinical testing could significantly impact our product development costs and commercial return potential, and could
result in the need for additional financing.
In
addition, if we change the regulatory pathway through which we intend to seek approval of any of our product candidates, or alter their
composition or method of manufacturing, we may have to conduct additional clinical trials, which may delay our ability to submit a marketing
application for the product. Even if we or our collaboration partners were to obtain approval for any of our product candidates, regulatory
agencies may limit the scope of such approval for fewer or more limited indications than we request, may grant approval contingent on
the completion of costly additional clinical trials or may approve a product candidate with a label that does not include the labeling
claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing could harm the commercial
prospects for our product candidates.
The
results of previous clinical trials may not be predictive of future results, and the results of our future clinical trials may not satisfy
the requirements of the FDA, EMA or other foreign regulatory agencies.
Clinical
failure can occur at any stage of clinical development. Clinical trials may produce negative or inconclusive results, and we or any of
our current and future collaborators may decide, or regulators may require us, to conduct additional clinical or preclinical testing.
We will be required to demonstrate with substantial evidence through well-controlled clinical trials that our product candidates are
as safe and effective for use for a specific indication(s) and in a specific patient population (s) before we can seek regulatory approvals
for their commercial sale. Success in early clinical trials does not mean that future larger registration clinical trials will be successful
because product candidates in later-stage clinical trials may fail to demonstrate safety and efficacy to the satisfaction of the FDA,
EMA and other foreign regulatory agencies despite having progressed through initial clinical trials. Product candidates that have shown
promising results in early clinical trials may still fail in subsequent confirmatory clinical trials. Similarly, the outcome of preclinical
testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial
do not necessarily predict final results. A number of companies in the pharmaceutical industry, including those with greater resources
and experience than us, have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier
clinical trials In addition, the design of a clinical trial can determine whether its results will support approval of a product and
flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. we may be unable to design
and execute a clinical trial to support regulatory approval. In some instances, there can be significant variability in safety or efficacy
results between different trials of the same product candidate due to numerous factors, including but not limited to changes in trial
protocols, differences in size and type of the patient populations, adherence to the dosing regimen and the rate of dropout among clinical
trial participants.
Further,
our product candidates may not be approved even if they achieve their primary endpoints in Phase 3 clinical trials or registration
trials. The FDA, EMA and other foreign regulatory agencies may disagree with our trial design and our interpretation of data from preclinical
studies and clinical trials. In addition, any of these regulatory authorities may change the requirements for the approval of a product
candidate even after reviewing and providing comments or advice on a protocol for a Phase 3 clinical trial that has the potential
to result in FDA or other agencies’ approval. We initially intend to seek approval for bevacizumab-anew for the treatment of metastatic
colorectal cancer and metastatic lung cancer. our plans to extrapolate to all other indications in the approved product labeling of the
reference product based on the sensitive population agreed by the FDA and EMA in the confirmatory clinical study. During review of the
registration application, our justification for the extrapolation may not be accepted. Any of the regulatory authorities may approve
a product candidate for fewer indications than we request or may grant approval contingent on the performance of costly post-marketing
clinical trials. In addition, the FDA, EMA and other foreign regulatory agencies may not approve the additional indication extrapolations
that we believe would be necessary or desirable for the successful commercialization of our product candidates.
Our
product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval,
limit the commercial profile of an approved label or result in significant negative consequences following marketing approval.
As
with most pharmaceutical products, use of our product candidates could be associated with side effects or adverse events, which can vary
in severity and frequency. Side effects or adverse events associated with the use of our product candidates may be observed at any time,
including in clinical trials or when a product is commercialized. Undesirable side effects caused by our product candidates could cause
us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or
denial of regulatory approval by the FDA or foreign authorities. Additionally, results of our studies could reveal a high and unacceptable
severity and prevalence of side effects, toxicity or other safety issues, and could require us to perform additional studies or halt
development or sale of these product candidates or expose us to product liability lawsuits that will harm our business. In such an event,
We may be required by regulatory agencies to conduct additional animal or human studies regarding the safety and efficacy of our product
candidates that we have not planned or anticipated or our studies could be suspended or terminated, and the FDA or comparable foreign
regulatory authorities could order us to cease further development of or deny or withdraw approval of our product candidates for any
or all targeted indications. There can be no assurance that we will resolve any issues related to any product-related adverse events
to the satisfaction of the FDA or any other regulatory agency in a timely manner, if ever, which could harm our business, prospects and
financial condition.
Additionally,
if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused
by such products, a number of potentially significant negative consequences could result.
If
we receive approval, regulatory agencies requires that we report certain information about adverse medical events.
The
timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event.
We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have
become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that
is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA, EMA or other
foreign regulatory agencies could take action including but not limited to criminal prosecution, the imposition of civil monetary penalties,
seizure of our products or delay in approval or clearance of future products.
If
other biosimilars of bevacizumab (Avastin) or rituximab-anew (Rituxan/Mabthera) are determined to be interchangeable and our biosimilar
product candidates for these reference products are not, our business would suffer.
The
FDA or other relevant regulatory authorities may determine that a proposed biosimilar product is “interchangeable” with a
reference product, meaning that the biosimilar product may be substituted for the reference product without the intervention of the healthcare
provider who prescribed the reference product, if the application includes sufficient information to show that the product is biosimilar
to the reference product and that it can be expected to produce the same clinical result as the reference product in any given patient.
If the biosimilar product may be administered more than once to a patient, the applicant must demonstrate that the risk in terms of safety
or diminished efficacy of alternating or switching between the biosimilar product and the reference product is not greater than the risk
of using the reference product without such alternation or switch. To make a final determination of biosimilarity or interchangeability,
regulatory authorities may require additional confirmatory information beyond what our plans to initially submit in our applications
for approval, such as more in-depth analytical characterization, animal testing or further clinical trials. Provision of sufficient information
for approval may prove. difficult and expensive.
We
cannot predict whether any of our biosimilar product candidates will meet regulatory authority requirements for approval as a biosimilar
product or as an interchangeable product in any jurisdiction. Furthermore, legislation governing interchangeability could differ by jurisdiction
on a state or national level worldwide.
If
product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization
of our current or future product candidates, and our insurance coverage may not be sufficient to satisfy any liability that may arise.
Drug-related
side effects could affect patient recruitment for clinical trials, the ability of enrolled patients to complete our studies or result
in potential product liability claims. We may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts
to protect us against losses due to liability. A successful product liability claim or series of claims brought against us could negatively
impact our results of operations and business. In addition, regardless of merit or eventual outcome, product liability claims may result
in impairment of our business reputation, withdrawal of clinical trial participants, costs due to related litigation, distraction of
management’s attention from our primary business, initiation of investigations by regulators, substantial monetary awards to patients
or other claimants, the inability to commercialize our product candidates and decreased demand for our product candidates, if approved
for commercial sale. Furthermore, we may also not be able to take advantage of limitations on product liability lawsuits that apply to
generic drug products, which could increase our exposure to liability for products deemed to be dangerous or defective.
Even
if we obtain regulatory approval for a product candidate, our products will remain subject to regulatory scrutiny.
If
our product candidates are approved, they will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging,
storage, advertising, promotion, sampling, record- keeping, conduct of post-marketing studies and submission of safety, efficacy and
other post-market information, including both federal and state requirements in the United States and requirements of comparable
foreign regulatory authorities. Manufacturers and manufacturing facilities are required to comply with extensive FDA, and comparable
foreign regulatory authority, requirements, including ensuring that quality control and manufacturing procedures conform to current Good
Manufacturing Practices, or cGMP, regulations. As such, we will be subject to continual review and inspections to assess compliance with
cGMP and adherence to commitments made in any non-disclosure agreement, BLA or marketing authorization application, or MAA. Accordingly,
we and our collaborators and suppliers must continue to expend time, money and effort in all areas of regulatory compliance, including
manufacturing, production and quality control.
If
a regulatory agency discovers previously unknown problems with an approved product, they may impose restrictions on that product.
If
we fail to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may, among other things:
| ● | issue
untitled and warning letters; |
| ● | impose
civil or criminal penalties; |
| ● | suspend
or withdraw regulatory approval; |
| ● | suspend
any of our ongoing clinical trials; |
| ● | refuse
to approve pending applications or supplements to approved applications submitted by us; |
| ● | impose
restrictions on our operations, including closing our manufacturing facilities; or |
| ● | seize
or detain products or require a product recall. |
If
regulatory sanctions are applied or if regulatory approval is withdrawn, the value of our company and our operating results will be negatively
impacted.
We
face intense competition and rapid technological change and the possibility that our competitors may develop therapies that are similar,
more advanced or more effective or less expensive than ours.
We
expect to enter highly competitive pharmaceutical markets. Successful competitors in the pharmaceutical markets have demonstrated the
ability to effectively discover, obtain patents, develop, test and obtain regulatory approvals for products, as well as an ability to
effectively commercialize, market and promote approved products. Numerous companies, universities and other research institutions are
engaged in developing, patenting, manufacturing and marketing of products competitive with those that we are developing. Many of these
potential competitors are large, experienced pharmaceutical companies that enjoy significant competitive advantages, such as substantially
greater financial, research and development, manufacturing, personnel and marketing resources. These companies also have greater brand
recognition and more experience in conducting preclinical testing and clinical trials of product candidates and obtaining FDA and other
regulatory approvals of products.
We
have competitors both in the United States and internationally, including major multinational pharmaceutical companies, specialty
pharmaceutical companies and biotechnology companies. Some of the pharmaceutical and biotechnology companies we expect to compete with
include, for example, Sandoz International GmbH, or Sandoz, Hospira, Inc., or Hospira, Amgen Inc., Pfizer Inc., Boehringer Ingelheim
GmbH, or Boehringer, Teva Pharmaceutical Industries, Ltd., Samsung Bioepis, Ltd. (a Merck/Biogen/Samsung biosimilar venture) and Hanwha
Chemical Corporation, as well as other smaller companies such as Coherus Biosciences, Inc. and Celltrion, Inc. At least four such competitors
have already obtained regulatory approval of, and have been marketing for several years their own biosimilar bevacizumab (Avastin) products.
Similarly, there are at least three approved rituximab biosimilars that have been on the market for several years. We will not be able
to obtain regulatory approval of either of its biosimilar product candidates for several more years (if ever) and by that time there
may be even more approved competing bevacizumab and rituximab biosimilar products on the market. which could materially harm our ability
to gain market share.
Many
of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff
and experienced marketing and manufacturing organizations.
As
a result, these companies may obtain regulatory approval more rapidly than we are able to and may be more effective in selling and marketing
their products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements
with large, established companies. Our competitors may also succeed in developing, acquiring or licensing on an exclusive basis, products
that are more effective or less costly than any product candidate that we may develop; they may also obtain patent protection that could
block our products; and they may obtain regulatory approval, product commercialization and market penetration earlier than we do.
If
an improved version of a reference product, such as Avastin, is developed or if the market for the reference product significantly declines,
sales or potential sales of our biosimilar product candidates may suffer.
Other
companies may develop improved versions of a reference product or the innovator applicant may change the reference product formulation
as part of a life cycle extension strategy and may obtain regulatory approval of the improved version under a new or supplemental Biologics
License Application (BLA) filed with the applicable regulatory authority. If a competitor succeeds in obtaining an approval of an improved
biologic product, it may capture a significant share of the collective market in the applicable jurisdiction and significantly reduce
the potential size of the market for our biosimilar product candidates. In addition, the improved product may be protected by additional
patent rights that may subject our follow-on biosimilar product to claims of infringement.
We
currently have no marketing and sales organization. If we are unable to establish sales and marketing capabilities in jurisdictions for
which we choose to retain commercialization rights, we may be unable to generate any revenue.
To
successfully commercialize any products that may result from our development programs, we will need to develop marketing and sales capabilities,
either on our own or with others. If our product candidates receive regulatory approval, we intend to establish a sales and marketing
organization with technical expertise and supporting distribution capabilities to commercialize our product candidates in major markets
where we may choose to retain commercialization rights. Doing so will be expensive, difficult and time-consuming. Any failure or delay
in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of our
products.
Further,
given our lack of prior experience in marketing and selling biosimilar or gene therapy products, our initial estimate of the size of
the required sales force may be materially more or less than the size of the sales force actually required to effectively commercialize
our product candidates. As such, we may be required to hire substantially more sales representatives and medical support liaisons to
adequately support the commercialization of our product candidates or e may incur excess costs as a result of hiring more sales representatives
than necessary.
We
may need to enter into agreements with other companies that can provide capabilities and funds for the development and commercialization
of our product candidates. If we are unsuccessful in forming or maintaining these agreements on favorable terms, our business could be
harmed.
Because
we have limited capabilities for late-stage product development, manufacturing, sales, marketing and distribution, we have found it necessary
to enter into agreements with other companies. Please see Section entitled “Intellectual Property — Our Licenses and Technology”.
In
the future, we may also find it necessary to enter into other agreements or joint ventures with major pharmaceutical companies to jointly
develop and/or commercialize its product candidates. If we are unable to secure or maintain such agreements we may not have the capabilities
necessary to continue or complete development of our product candidates and bring them to market, which may have an adverse effect on
our business.
In
addition to commercialization capabilities, we may depend on our agreements with other companies to provide our additional funding for
development and potential commercialization of our product candidates. We may not be able to obtain funding on favorable terms, and if
we is not successful in doing so, we may not have sufficient funds to develop a particular product candidate internally or to bring product
candidates to market. As a result, our business and operating results may be harmed.
The
third-party coverage and reimbursement status of newly approved products is uncertain. Failure to obtain or maintain adequate coverage
and reimbursement for new or current products could limit our ability to market those products and decrease our ability to generate revenue.
Pricing,
coverage and reimbursement of our biosimilar and gene therapy product candidates, if approved, may not be adequate to support our commercial
infrastructure. Our per-patient prices may not be sufficient to recover our development and manufacturing costs and potentially achieve
profitability. The availability and adequacy of coverage and reimbursement by governmental and private payors are essential for most
patients to be able to afford expensive treatments such as ours, if approved. Accordingly, sales of our product candidates will depend
substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid for by health maintenance,
managed care, pharmacy benefit and similar healthcare management organizations or reimbursed by government authorities, private health
insurers and other third- party payors. If coverage and reimbursement are not available, or are available only at insufficient levels,
We may not be able to successfully commercialize our product candidates.
There
is significant uncertainty related to third-party coverage and reimbursement of newly approved products. In the United States, third-party
payors, including private and governmental payors such as the Medicare and Medicaid programs, play an important role in determining the
extent to which new drugs and biologics will be covered and reimbursed. The Medicare and Medicaid programs increasingly are used as models
for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. It is
difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our biosimilar
product candidates, if approved. In addition, in the United States, no uniform policy of coverage and reimbursement for biologics
or gene therapy exists among third-party payors. Therefore, coverage and reimbursement can differ significantly from payor to payor.
As a result, the process for seeking favorable coverage determinations often is time-consuming and costly and may require us to provide
scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement
will be obtained. our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private
payors for any approved products could have a material adverse effect on our operating results, our ability to raise capital needed to
commercialize products and our overall financial condition.
Our
biosimilar product candidates, if approved, will face price competition from both the respective reference products and other biosimilars.
Successful
competitors in the biosimilar market will likely have the ability to effectively compete on price through payors and their third-party
administrators who exert downward pricing pressure. It is possible our competitors’ compliance with price discounting demands in
exchange for market share could exceed our capacity to respond in kind and reduce market prices beyond our expectations. In addition,
the RPS may compete effectively on price and limit our ability to accrue market share. Such practices may limit our and our collaboration
partners’ ability to increase market share and will also impact profitability.
Enrollment
and retention of patients in clinical trials is an expensive and time-consuming process and could be made more difficult or rendered
impossible by multiple factors outside our control.
We
may encounter delays in enrolling, or be unable to enroll, a sufficient number of patients to complete any of our clinical trials, and
even once enrolled We may be unable to retain a sufficient number of patients to complete any of our trials. Patient enrollment and retention
in clinical trials depends on many factors, including the size of the patient population, the nature of the trial protocol, the effectiveness
of our patient recruitment efforts, the existing body of safety and efficacy data with respect to the study candidate, the perceived
risks and benefits of gene therapy approaches for the treatment of neurological diseases, the number and nature of competing existing
treatments for our target indications, the number and nature of ongoing trials for other product candidates in development for our target
indications, perceived risk of the delivery procedure, patients with pre-existing conditions that preclude their participation in any
trial, the proximity of patients to clinical sites and the eligibility criteria for the study. Furthermore, the negative results we have
reported in its biosimilar product clinical trials to date and any other negative results we may report in clinical trials of any of
our gene therapy or biosimilar product candidates in the future may make it difficult or impossible to recruit and retain patients in
other clinical trials of those product candidates. Similarly, negative results reported by our competitors about their product candidates
may negatively affect patient recruitment in our clinical trials. Delays or failures in planned patient enrollment or retention may result
in increased costs, program delays or both, which could have a harmful effect on our ability to develop our product candidates or could
render further development impossible. In addition, we expect to rely on contract research organizations (CROs) and clinical trial sites
to ensure proper and timely conduct of our future clinical trials and, while we intend to enter into agreements governing their services,
we will be limited in our ability to control their actual performance.
Our
AAV-based gene therapy product candidates and our lentiviral-based gene therapy product candidate are based on new gene transfer technology,
which makes it difficult to predict the time and cost of product candidate development and of subsequently obtaining regulatory approval.
The
use of gene therapy in the treatment of Alzheimer’s and ALS is new but promising. We may experience problems or delays in developing
new gene therapy product candidates and such problems or delays may cause unanticipated costs, and such development problems may not
be solvable. we may also experience delays in developing a sustainable, reproducible and scalable manufacturing process or transferring
that process for our gene therapy product candidates from their current manufacturers, which may prevent us from completing our clinical
studies or commercializing our products on a timely or profitable basis, if at all.
In
addition, the clinical trial requirements of the FDA and other foreign regulatory authorities and the criteria these regulators use to
determine the safety and efficacy of a product candidate vary according to the type, complexity, novelty and intended use and market
of such product candidates. The regulatory approval process for novel product candidates such as ours can be more expensive and take
longer than for other, better known or more extensively studied product candidates. To date, only a limited number of gene therapies
have received marketing authorization from the FDA or foreign regulatory authorities.
Since
August 2017, the FDA has approved 32 cell and gene therapy products including two that utilized a lentiviral-based vector. It is
difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our gene therapy product candidates
in either the United States, or other major markets or how long it will take to commercialize our gene therapy product candidates,
if any are approved. Approvals by foreign regulatory authorities may not be indicative of what the FDA may require for approval, and
vice versa.
Clinical
Development of our gene therapy candidates will require the development and use of new in vitro assays that can detect and measure different
isoforms and metabolites of Klotho.
Our
R&D program for our gene therapy products will requires us to develop and validate one or more assays to separate and individually
measure the three isoforms (s-KL, m-KL, p-KL), and the two metabolites (KL1and KL2) of Klotho. If we encounter delays or are unable to
successfully develop and obtain necessary regulatory approvals for such assays, we may be unable to develop our gene therapy candidates
on a timely basis or at all.
Regulatory
requirements governing gene therapy products have changed frequently and may continue to change in the future.
The
FDA has established the Office of Tissues and Advanced Therapies within its Center for Biologics Evaluation and Research (“CBER”)
to consolidate the review of gene therapy and related products, and has established the Cellular, Tissue and Gene Therapies Advisory
Committee to advise the CBER in its review. If we were to engage an NIH-funded institution, to conduct a clinical trial, that institution’s
institutional biosafety committee as well as its IRB would need to review the proposed clinical trial to assess the safety of the trial.
In addition, adverse developments in clinical trials of gene therapy products conducted by others may cause the FDA or other oversight
bodies to change the requirements for approval of any of our gene therapy product candidates. Similarly, foreign regulatory authorities
may issue new guidelines concerning the development and marketing authorization for gene therapy medicinal products and require that
e comply with these new guidelines.
The
FDA, NIH and the EMA have each expressed interest in further regulating biotechnology, including gene therapy and genetic testing.
Agencies
at both the federal and state level in the United States, as well as the U.S. Congressional committees and other governments
or governing agencies, have expressed interest in further regulating the biotechnology industry. Such action may delay or prevent commercialization
of some or all of our gene therapy product candidates. These regulatory review committees and advisory groups and any new guidelines
they promulgate may lengthen the regulatory review process, require us to perform additional studies, increase our development costs,
lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these product candidates
or lead to significant post- approval limitations or restrictions. As we advance our gene therapy product candidates, we will be required
to consult with these regulatory and advisory groups and comply with applicable guidelines. If e fail to do so, we may be required to
delay or discontinue development of certain of our gene therapy product candidates. These additional processes may result in a review
and approval process that is longer than we otherwise would have expected. Delay or failure to obtain, or unexpected costs in obtaining,
the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient product revenue,
and our business, financial condition, results of operations and prospects would be materially and adversely affected.
Even
if we obtain FDA approval for our gene therapy product candidates in the United States, we may never obtain approval for or commercialize
them in any other jurisdiction, which would limit our ability to realize their full market potential.
In
order to market any products in any particular jurisdiction, we must establish and comply with numerous and varying regulatory requirements
on a country-by-country basis regarding safety and efficacy. Approval by the FDA in the United States does not ensure approval by
regulatory authorities in other countries or jurisdictions. In addition, clinical trials conducted in one country may not be accepted
by regulatory authorities in other countries, and regulatory approval in one country does not guarantee regulatory approval in any other
country. Approval processes vary among countries and can involve additional product testing and validation and additional administrative
review periods. Seeking foreign regulatory approval could result in difficulties and costs for us and require additional nonclinical
studies or clinical trials which could be costly and time consuming. Regulatory requirements can vary widely from country to country
and could delay or prevent the introduction of our products in those countries.
Even
if our product candidates receive marketing approval, they may fail to achieve market acceptance by physicians, patients, third-party
payors or others in the medical community necessary for commercial success.
Even
if our product candidates receive marketing approval, they may nonetheless fail to gain sufficient market acceptance by physicians, patients,
third-party payors and others in the medical community, including due to the novelty of gene therapy products in general. If they do
not achieve an adequate level of acceptance, we may not generate significant product revenues and become profitable. The degree of market
acceptance for our product candidates, if approved for commercial sale, will depend on a number of factors, including but not limited
to:
| ● | the
efficacy and potential advantages compared to alternative treatments; |
| ● | the
effectiveness of sales and marketing efforts; |
| ● | the
cost of treatment in relation to alternative treatments; |
| ● | our
ability to offer our products for sale at competitive prices; |
| ● | the
willingness of the target patient population to try new therapies and of physicians to prescribe
these therapies; |
| ● | the
ethical, social and legal concerns about gene therapy; |
| ● | the
availability of third-party coverage and adequate reimbursement and patients’ willingness
to pay for our products in the absence of such coverage and adequate reimbursement; |
| ● | the
prevalence and severity of any side effects; and |
| ● | any
restrictions on the use of our product together with other medications. |
The
failure of any of our product candidates, if approved, to find market acceptance would harm our business and could require us to seek
additional financing.
Negative
public opinion and increased regulatory scrutiny of gene therapy and genetic research may damage public perception of our product candidates
or adversely affect our ability to conduct our business or obtain regulatory approvals for our product candidates.
Gene
therapy remains a novel technology, with only a limited number of gene therapy products approved to date. Public perception may be influenced
by claims that gene therapy is unsafe, unethical or immoral, and gene therapy may not gain the acceptance of the public or the medical
community. In particular, our success will depend upon the comfort level of physicians to prescribe our product candidates, in lieu of,
or in addition to, existing or standard treatments they are already familiar with and for which greater clinical data may be available.
More
restrictive government regulations or negative public opinion would have a negative effect on our business or financial condition and
may delay or impair the development and commercialization of our gene therapy product candidates. Earlier gene therapy trials led to
several well-publicized adverse events, including cases of leukemia and death seen in such trials using earlier generation vectors. In
addition, there is the potential risk of delayed adverse events following exposure to gene therapy products due to persistent biological
activity of the genetic material or other components of products used to carry the genetic material. Gene therapy that targets a gene
that is, or that near to another gene, that is important in cell growth or division could potentially enhance the risk of malignant transformation.
If our vectors demonstrate such an effect, we may decide or be required to halt or delay further clinical development of our AAV-based
or lentiviral-based product candidates.
Adverse
events in our clinical studies, even if not ultimately attributable to our product candidates (such as the many adverse events that typically
arise from the conditioning process), or adverse events in other lentiviral gene therapy trials, and the resulting publicity, could result
in increased governmental regulation, unfavorable public perception, potential regulatory delays in the testing or approval of our product
candidates, stricter labeling requirements for those product candidates that are approved and a decrease in demand for any such product
candidates.
Increasing
demand for compassionate use or expanded access of our unapproved therapies could negatively affect our reputation and harm our business.
We
are developing our gene therapy product candidates for life-threatening illnesses for which there are currently limited to no available
therapeutic options. It is possible for individuals or groups to target companies with disruptive social media campaigns related to a
request for access to unapproved drugs for patients with significant unmet medical need. If we experience a similar social media campaign
regarding our decision to provide or not provide our product candidates under an expanded access corporate policy, our reputation may
be negatively affected and our business may be harmed.
Recent
media attention to individual patients’ expanded access requests has resulted in the introduction of legislation at the local and
national level referred to as “Right to Try” laws, such as the Right to Try Act, which are intended to give patients access
to unapproved therapies. New and emerging legislation regarding expanded access to unapproved drugs for life-threatening illnesses could
negatively impact our business in the future.
A
possible consequence of both activism and legislation in this area is the need for us to initiate an unanticipated expanded access program
or to make our product candidates more widely available sooner than anticipated. We are a small company with limited resources and unanticipated
trials or access programs could result in diversion of resources from our primary goals.
In
addition, some patients who receive access to unapproved drugs through compassionate use or expanded access programs have life-threatening
illnesses and have exhausted all other available therapies. The risk for serious adverse events in this patient population is high which
could have a negative impact on the safety profile of our product candidates could cause significant delays or an inability to successfully
commercialize our product candidates, which could materially harm our business. If we were to provide patients with our product candidates
under an expanded access program, We may in the future need to restructure or pause ongoing compassionate use and/or expanded access
programs in order to perform the controlled clinical trials required for regulatory approval and successful commercialization of our
product candidates, which could prompt adverse publicity or other disruptions related to current or potential participants in such programs.
Risks
Related to Our Reliance on Third Parties
We
will rely on third parties to manufacture and quality control test our product candidates, to conduct our preclinical and clinical trials,
and perform other tasks for us.
We
plan to rely upon contract research organizations (CROs) and contract process development and manufacturing organizations (CDMOs) to
monitor and manage manufacturing supply and collect and manage data for our ongoing preclinical and clinical programs and we will rely
on these parties for quality product supply, logistics, and execution of our preclinical and clinical trials and regulatory submissions,
and we can only control certain aspects of their activities. Nevertheless, we re responsible for ensuring that each of our studies is
conducted in accordance with the applicable protocol, legal, regulatory and scientific requirements and standards and our reliance on
the CROs does not relieve us of our regulatory responsibilities. If we, or any of our CROs/CDMOs, service providers or investigators
fails to comply with applicable regulations or GCPs, the data generated in our preclinical and clinical trials may be deemed unreliable
and the FDA, EMA or comparable foreign regulatory authorities may require us to perform additional preclinical and clinical trials before
approving our marketing applications. Failure to comply by any of the participating parties or ourselves with these regulations may require
us to repeat clinical trials, which would delay the regulatory approval process.
We
expect to depend on third parties for the commercialization of our biosimilar and gene therapy product candidates in certain major markets
outside the United States, and their failure to commercialize in those markets could harm our business and operating results.
We
will try to maximize the value of our product pipeline by retaining development and commercialization rights in the United States
and continuing to selectively out-license to ex-U.S. markets. Accordingly, we will need to identify third-parties and then negotiate
the terms of the development and commercialization agreements for major ex-U.S. markets, such as the EU and Japan. We may not be
successful in identifying contract counterparties, and we may not be able to reach agreements with such parties on terms that are as
favorable to our company as we would anticipate. If these entities fail to exercise commercially reasonable efforts to market and sell
our products in their respective licensed jurisdictions or are otherwise ineffective in doing so, our business will be harmed and we
may not be able to adequately remedy the harm through negotiation, litigation, arbitration or termination of the license agreements.
We
and RLS, and our gene therapy CDMO, are subject to significant regulation with respect to manufacturing our product candidates. RLS and
the CDMO’s manufacturing facilities may not continue to meet regulatory requirements or may not be able to meet supply demands.
Components
of a finished therapeutic product approved for commercial sale or used in late-stage clinical trials must be manufactured in accordance
with cGMP and other applicable regulations. Poor control of production processes can lead to the introduction of contaminants or to inadvertent
changes in the properties or stability of our NEW’s product candidates that may not be detectable in final product testing. we
must supply all necessary documentation in support of a BLA or MAA on a timely basis and must adhere to GLP and cGMP regulations enforced
by the FDA and other regulatory agencies through their facilities inspection program. In addition, the regulatory authorities may, at
any time, audit or inspect our manufacturing facility or our associated quality systems for compliance with the regulations applicable
to the activities being conducted. If our facilities do not pass a pre-approval facility inspection, regulatory approval of the products
may not be granted or may be substantially delayed until any violations are corrected to the satisfaction of the regulatory authority,
if ever. If RLS or other CDMOs fail to maintain regulatory compliance, the FDA or other applicable regulatory authority can impose regulatory
sanctions including, among other things, refusal to approve a pending application for a new biologic product, withdrawal of an approval
or suspension of production. As a result, our business, financial condition and results of operations may be harmed.
The
regulatory authorities also may, at any time following approval of a product for sale, audit our manufacturing facilities. If any such
inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable
regulations occurs independent of such an inspection or audit, the relevant regulatory authority may require remedial measures that may
be costly and time-consuming for us to implement and that may include the temporary or permanent suspension of a clinical trial or commercial
sales or the temporary or permanent closure of our facility. Any such remedial measures could harm our business.
Risks
Related to Intellectual Property
If
we infringe or are alleged to infringe intellectual property rights of third parties, our business could be harmed. Third-party claims
of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our
commercial success depends in large part on avoiding infringement of the patents and proprietary rights of third parties. There have
been many lawsuits and other proceedings involving patent and other intellectual property rights in the pharmaceutical industry, including
patent infringement lawsuits, interferences, oppositions, interested parties review and reexamination proceedings before the U.S. Patent
and Trademark Office, or USPTO, and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent
applications, which are owned by third parties, exist in the fields in which we are developing product candidates. As the pharmaceutical
industry expands and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement
of the patent rights of third parties.
Our
research, development and commercialization activities may infringe or otherwise violate or be claimed to infringe or otherwise violate
patents owned or controlled by other parties. The companies that originated the products for which our intend to introduce biosimilar
versions, such as Genentech, Inc., or Roche, as well as other competitors (including other companies developing biosimilars) have developed
worldwide patent portfolios of varying sizes and breadth, many of which are in fields relating to our business, and it may not always
be clear to industry participants, including us, which patents cover various types of products, formulations, manufacturing processes
or methods of use.
Third
parties may assert that we are employing their proprietary technology without authorization.
There
may be third-party patents or patent applications with claims to compositions, formulations, methods of manufacture or methods for treatment
related to the use or manufacture of our product candidates.
Moreover,
because patent applications can take many years to issue, there may be currently pending patent applications that may later result
in issued patents covering our product candidates. The existence of any patent with valid and enforceable claims covering one or more
of our product candidates could cause substantial delays in our ability to introduce such product candidate into the U.S. market
if the term of such patent extends beyond our desired product launch date.
Additionally,
we may face claims from non-practicing third-party entities that have no relevant product revenue and against whom our own patent portfolio
may have no deterrent effect. In addition, the scope of patent claims is subject to interpretation by the courts. If we are sued for
patent infringement, we would need to demonstrate that our product candidates, methods of manufacture or methods of use either do not
infringe the asserted patent claims or that the claims are invalid and/or unenforceable, and we may not be successful. Even if we are
successful in litigation, we may incur substantial costs and the time and attention of our management and scientific personnel could
be diverted, which could harm our business. In addition, we may not have sufficient resources to bring these actions to a successful
conclusion.
Third
parties could bring claims against us that would cause us to incur substantial expenses and, if successful against us, could cause us
to pay substantial monetary damages.
The
outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. If a patent
infringement suit were brought against us, we could be forced to stop or delay research, development, manufacturing or sales of the product
or product candidate that is the subject of the suit. Ultimately, we could be prevented from commercializing a product or be forced to
cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we are unable to enter
into licenses on commercially acceptable terms or at all. If, as a result of patent infringement claims or to avoid potential claims,
we choose or are required to seek licenses from third parties, these licenses may not be available on acceptable terms or at all. Even
if we are able to obtain a license, the license may obligate us to pay substantial license fees or royalties or both, and the rights
granted to us might be nonexclusive, which could result in our competitors gaining access to the same intellectual property.
Moreover,
parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further
develop and commercialize one or more of our product candidates. Defense of these claims, regardless of their merit, would likely involve
substantial litigation expense and would likely be a substantial diversion of employee resources from our business. In the event of a
successful claim of infringement against our, our may, in addition to being blocked from the market, have to pay substantial monetary
damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products
or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
We
may not identify relevant patents or may incorrectly interpret the relevance, scope or expiration of a patent, which might adversely
affect our ability to develop and market our products.
The
scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution
history.
Further,
the identification of all patents and their expiration dates relevant to the production and sale of a reference product is extraordinarily
complex and requires sophisticated legal knowledge in the relevant jurisdiction. It may be impossible to identify all patents in
all jurisdictions relevant to a marketed product. we may not identify all relevant patents, or incorrectly determine their expiration
dates, which may negatively impact our ability to develop and market our products. Our failure to identify and correctly interpret relevant
patents may negatively impact our ability to develop, market and commercialize our products.
We
may become involved in lawsuits to protect or enforce any future patents, which could be expensive, time-consuming and unsuccessful.
We
may discover that competitors are infringing patents we have in-licensed or that may be granted to us in the future. Expensive and time-consuming
litigation may be required to enforce our in-licensed or future patents. If we or one of our collaboration partners were to initiate
legal proceedings against a third party to enforce a patent covering one of our product candidates, the defendant could counterclaim
that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant
counterclaims alleging invalidity and/or unenforceability are commonplace. The outcome following legal assertions of invalidity and unenforceability
is unpredictable, and there is a risk that a court will decide that a patent of ours is invalid or unenforceable, in whole or in part,
and that we do not have the right to stop the other party from using the invention at issue. There is also a risk that, even if the validity
of such patents is upheld, the court will construe the patent’s claims narrowly and decide that we do not have the right to stop
the other party from using the invention at issue on the grounds that our patent claims do not cover the invention. An adverse outcome
in a litigation or proceeding involving our patents could limit our ability to assert our patents against those parties or other competitors,
and may curtail or preclude our ability to exclude third parties from making and selling similar or competitive products. Any of these
occurrences could adversely affect our competitive business position, business prospects and financial condition.
Furthermore,
because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some
of our confidential information could be compromised by disclosure during any litigation we initiate to enforce our in-licensed or future
patents. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If
securities analysts or investors perceive these results to be negative, it could have a negative impact on the price of our common stock.
Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims,
which typically last for years before they are concluded. Even if we ultimately prevail in such claims, the monetary cost of such
litigation and the diversion of the attention of our management and scientific personnel could outweigh any benefit we receive as a result
of the proceedings.
We
may be unable to obtain and maintain effective patent rights for our product candidates or any future product candidates.
We
rely upon a combination of licensed patents, trade secret protection and confidentiality agreements to protect our intellectual property
related to our product candidates and development programs. Our ability to enjoy any competitive advantages afforded by our intellectual
property depends in large part on our ability to obtain and maintain patents and other intellectual property protection in the United States
and in other countries with respect to various proprietary elements of our product candidates, such as, for example, Our product formulations
and processes for manufacturing our product candidates and our ability to maintain and control the confidentiality of our trade secrets
and confidential information critical to our business.
Although
it has not filed patents to date, we plan to seek to protect our proprietary position by filing patent applications in the United States
and abroad related to our products that are important to our business. This process is expensive and time-consuming, and we may not be
able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible
that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection.
There is no guarantee that any patent application we file will result in an issued patent having claims that protect our products; and,
as a result, we may not be able to effectively prevent others from commercializing competitive products. Additionally, while the basic
requirements for patentability are similar across jurisdictions, each jurisdiction has its own specific requirements for patentability.
We cannot guarantee that we will obtain identical or similar patent protection covering our products in all jurisdictions where we files
patent applications.
The
patent positions of biopharmaceutical companies generally are highly uncertain and involve complex legal and factual questions for which
legal principles remain unresolved. As a result, the claims that cover our product candidates in the United States may not provide
the same degree of protection as patent claims in other foreign countries. There is no assurance that all potentially relevant prior
art relating to our patents and patent applications has been found, considered or cited during patent prosecution, which can be used
to invalidate a patent or prevent a patent from issuing from a pending patent application. Even if patents do successfully issue, and
even if such patents cover our product candidates, third parties may challenge their validity, enforceability or scope, which may result
in such patent claims being narrowed, found unenforceable or invalidated. Furthermore, even if they are unchallenged, our patents may
not adequately protect our intellectual property, provide exclusivity for our product candidates or prevent others from designing around
our claims. Any of these outcomes could impair our ability to prevent competitors from using the technologies claimed in any patents
issued to us, which may have an adverse impact on our business.
Patents
granted by the European Patent Office may be opposed by any person within nine months from the publication of their grant and, in
addition, may be challenged before national courts at any time. Furthermore, even if they are unchallenged, our patents and patent applications
may not adequately protect our intellectual property or prevent others from designing around our claims. If the breadth or strength of
protection provided by the patents and patent applications we hold, license or pursue with respect to our product candidates is threatened,
it could threaten our ability to prevent third parties from using the same technologies that we use in our product candidates.
Obtaining
and, once patents are obtained, maintaining our patent protection will depend on compliance with various procedural requirements, document
submissions, fee payment and other requirements imposed by governmental patent agencies. our patent protection could be reduced or eliminated
for non-compliance with these requirements.
The
USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other
provisions during the patent process. In certain cases, an inadvertent lapse can be cured by payment of a late fee or by other means
in accordance with the applicable rules. However, there are situations in which noncompliance can result in abandonment or lapse of a
patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event,
competitors might be able to enter the market earlier than would otherwise have been the case and it may materially affect our business.
We
may not be able to protect our intellectual property rights throughout the world.
Filing,
prosecuting, defending and enforcing intellectual property on product candidates in all countries throughout the world would be prohibitively
expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in
the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent
as federal and state laws in the United States. Further, licensing partners may choose not to file patent applications in certain
jurisdictions in which we may obtain commercial rights, thereby precluding the possibility of later obtaining patent protection in these
countries. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States
or importing products made using our inventions into the United States or other jurisdictions.
Risks
Related to Our Business Operations
We
may not be successful in our efforts to identify, develop or commercialize additional product candidates.
Although
a substantial amount of our effort will focus on the continued clinical testing, potential approval and commercialization of our existing
product candidates, the success of our business also depends upon our ability to identify, develop and commercialize additional product
candidates. Research programs to identify new product candidates require substantial technical, financial and human resources. We may
focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful. Our development
efforts may fail to yield additional product candidates suitable for clinical development and commercialization for a number of reasons,
including but not limited to the following:
| ● | We
may not be successful in identifying potential product candidates that pass our strict screening
criteria; |
| ● | We
may not be able to overcome technological hurdles to development or a product candidate may
not be capable of producing commercial quantities at an acceptable cost, or at all; |
| ● | We
may not be able to patent “natural” or known human gene compositions of matter
or use in treating or preventing diseases; |
| ● | We
may not be able to assemble sufficient resources to acquire or discover additional product
candidates; |
| ● | Our
product candidates may not succeed in preclinical or clinical testing; |
| ● | Our
potential product candidates may fail to show sufficient biosimilarity to reference molecules;
and |
| ● | Competitors
may develop alternatives that render our product candidates obsolete or less attractive or
the market for a product candidate may change such that a product candidate may not justify
further development. |
If
any of these events occur, we may be forced to abandon our development efforts for a program or programs or we may not be able to identify,
develop or commercialize additional product candidates, which would harm our business and could potentially cause our to cease operations.
We
may be subject, directly or indirectly, to federal and state healthcare laws and regulations, including fraud and abuse, false claims,
physician payment transparency and health information privacy and security laws. If we are unable to comply or have not fully complied
with such laws, we could face substantial penalties.
If
we obtain FDA approval for any of our product candidates and begins commercializing those products in the United States, our operations
may be directly or indirectly through our customers subject to various federal and state fraud and abuse laws, including without limitation,
the federal Anti-Kickback Statute, the federal False Claims Act and physician sunshine laws and regulations. These laws may impact, among
other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient data privacy and security
regulation by both the federal government and the states in which our conduct our business.
If
our operations are found to be in violation of any laws or any other governmental regulations that apply to us, we may be subject to
penalties, including civil and criminal penalties, damages, fines, exclusion from participation in government healthcare programs, such
as Medicare and Medicaid, imprisonment, disgorgement, contractual damages, reputational harm, diminished profits and future earnings,
and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our
results of operations. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel
resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may
be impaired.
If
we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur
costs that could harm our business.
Our
research, development and manufacturing activities and our third-party suppliers’ activities involve the controlled storage, use
and disposal of hazardous materials, including the components of our product candidates and other hazardous compounds. We and our suppliers
are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. In some
cases, these hazardous materials and various wastes resulting from their use are stored at our facilities pending their use and disposal.
We cannot eliminate the risk of contamination, which could cause an interruption of our commercialization efforts, research, development
and manufacturing efforts and business operations, and environmental damage resulting in costly clean-up and liabilities under applicable
laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we believe
that the safety procedures utilized by us for handling and disposing of these materials generally comply with the standards prescribed
by these laws and regulations, We cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury
from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources
and state or federal or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations.
Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict
the impact of such changes and cannot be certain of our future compliance. We do not currently carry biological or hazardous waste insurance
coverage.
General
Risks
We
are subject to a variety of stringent and changing privacy and data security laws, contractual obligations, self-regulatory schemes,
government regulation, and standards related to data privacy and security. The actual or perceived failure by us, our partners, or vendors
to comply with such obligations could harm our reputation, subject us to significant fines and liability, disrupt our clinical trials,
or otherwise adversely affect our business.
We
collect, receive, store, process, use, generate, transfer, disclose, make accessible, protect and share personal information and other
information, including information we collect about patients and healthcare providers in connection with clinical trials in the U.S. and
abroad, to operate our business, for legal and marketing purposes, and for other business-related purposes.
There
are numerous federal, state, local, and international laws, regulations, and guidance regarding privacy, and information use, storage
and security, the number and scope of which is changing, subject to differing applications and interpretations, and which may be inconsistent
or in conflict with other rules among jurisdictions. For example, US States have also begun to introduce more and more comprehensive
privacy legislation. The California Consumer Privacy Act of 2018, or CCPA, affords consumers expanded privacy protections.
Aspects of the CCPA and its interpretation and enforcement remain uncertain. The potential effects of the CCPA are far-reaching and may
require us to modify our data processing practices and policies and to incur substantial costs and expenses in an effort to comply.
We
are also subject to the terms of our privacy and security policies, representations, certifications, standards, publications and frameworks,
or Privacy Policies, and contractual obligations to third parties related to privacy, and information use, storage and security. While
we strive to comply with applicable data protection laws we may at times fail to do so, or may be perceived to have failed to do so.
Moreover, despite our efforts, we may not be successful in achieving compliance if our personnel, partners or vendors do not comply with
applicable laws, Privacy Policies and other obligations. In such event, such failure or perceived failure could: increase our compliance
and operational costs; expose us to regulatory scrutiny, actions, fines and penalties; result in reputational harm; interrupt or stop
clinical trials; result in litigation and liability; result in an inability to process personal information or to operate in certain
jurisdictions; and cause a material adverse impact to business operations or financial results.
If
our security measures are compromised now, or in the future, or the security, confidentiality, integrity or availability of, our information
technology, software, services, communications or data is compromised, limited or fails, it could result in material harm to our business.
In
the ordinary course of our business, we may collect, process and store proprietary, confidential and sensitive information, including
personal information (including health information), intellectual property, trade secrets, and proprietary business information owned
or controlled by ourselves or other parties. We may use third-party service providers and sub-processors to help we operate our business
and engage in use of data on our behalf. We may also share sensitive information with our partners or other third parties in conjunction
with our business. If we, our service providers, partners or other relevant third parties have experienced, or in the future experience,
any security incident(s) that result in, any data loss, deletion or destruction, unauthorized access to, loss of, unauthorized acquisition
or disclosure of, or compromise related to the security, confidentiality, integrity or availability of our information technology, software,
services, communications or data (or those of our service providers, partners or other relevant third parties), it may have a material
adverse effect on our business, including without limitation, regulatory investigations or enforcement actions, litigation, indemnity
obligations, negative publicity and financial loss. For example, the loss of clinical trial data from completed or ongoing or planned
clinical trials could result in delays in our regulatory approval efforts and could require us to incur substantial cost to recover or
reproduce such data.
In
addition, cyberattacks, malicious internet-based activity and online and offline fraud are prevalent and continue to increase. In addition
to traditional computer “hackers,” threat actors, software bugs, malicious code (such as viruses and worms), employee theft
or misuse, denial-of-service attacks (such as credential stuffing), and ransomware attacks, sophisticated nation-state and nation-state
supported actors now engage in attacks (including advanced persistent threat intrusions). We may also be the subject of phishing attacks,
viruses, malware installation, server malfunction, software or hardware failures, loss of data or other computer assets, adware or other
similar issues.
We
may be required to expend significant resources, fundamentally change business activities and practices, or modify our operations, including
our clinical trial activities or information technology, in an effort to protect against data breaches and to mitigate, detect, and remediate
actual and potential vulnerabilities. Applicable laws may require us to implement specific security measures or use industry-standard
or reasonable measures to protect against data breaches. While we have implemented security measures designed to protect against data
breaches there can be no assurance that our measures or those of our service providers, partners and other third parties, will be effective
in protecting against all data breaches and material adverse impacts that may arise from such breaches. The recovery systems, security
protocols, network protection mechanisms and other security measures that we (and the third parties that our works with) have integrated
into our platform, systems, networks and physical facilities, which are designed to protect against, detect and minimize data breaches,
may not be adequate to prevent or detect service interruption, system failure or data loss.
We
may not have adequate insurance coverage in the event of a Security Breach. We cannot assure that our existing coverage will be adequate
or otherwise protect us from or adequately mitigate liabilities or damages with respect to claims we may experience, or that such coverage
will continue to be available on acceptable terms or at all. The successful assertion of one or more large claims against us that exceeds
our available insurance coverage, or results in changes to our insurance policies (including premium increases or the imposition of large
deductible or co-insurance requirements), could have an adverse effect on our business. In addition, we cannot be sure that our existing
insurance coverage and coverage for errors and omissions will continue to be available on acceptable terms or that our insurers will
not deny coverage as to any future claim.
The
occurrence of the COVID-19 pandemic may negatively affect our operations depending on the severity and longevity of the pandemic.
The
COVID-19 pandemic is currently impacting countries, communities, supply chains and markets as well as the global financial markets. A
pandemic has resulted in social distancing, travel bans and quarantine which have limited access to our facilities, management, support
staff and professional advisors. These factors, in turn, may not only impact our operations, financial condition and demand for our goods
and services but our overall ability to react timely to mitigate the impact of this event. Also, it may hamper our efforts to comply
with our filing obligations with the Securities and Exchange Commission. Depending on the severity and longevity of the COVID-19 pandemic,
our business and shareholders may experience a significant negative impact. Currently, the COVID-19 pandemic has limited our ability
to move forward with our operations and has negatively affected our ability to timely comply with our ongoing filing obligations with
the Securities and Exchange Commission. We cannot offer any assurance as to our future financial results. You may lose your entire investment.
We
are a new business and as such, there is no assurance that we will be able to locate customers that could use our services in the future
and we may not be able to generate revenues in the future in a manner that will be sufficient for us to remain profitable.
Our
future profitability will be dependent upon if we can service clients and increase our client base. There can be no assurance that
we will ever increase our profitability.
Even
if we obtain future revenues sufficient to expand operations, increased operating expenses could adversely affect our ability to operate
in a profitable manner.
We
may in the future issue more shares that could cause a loss of control by our present management and current stockholders.
We
may issue further shares as consideration for the cash or assets or services out of our authorized, but unissued, common stock that would,
upon issuance, represent a majority of our voting power and equity. The result of such an issuance would be those new stockholders and
management would control the Company, and persons unknown could replace our management at that time. Such an occurrence would result
in a greatly reduced percentage of ownership of the Company by our current shareholders, which could present significant risks to investors.
The
elimination of personal liability of director and officers under Delaware law and the existence of indemnification rights held by our
directors, officers and employees may result in substantial expenses.
We
have agreed to indemnification of officers and director as provided by the Delaware General Corporation Law. The Delaware General Corporation
Law provides for the indemnification of our directors, officers, employees, and agents, under certain circumstances, against attorney’s
fees and other expenses incurred by them in any litigation to which they become a party arising from their association with or activities
on our behalf. We will also bear the expenses of such litigation for any of our directors, officers, employees, or agents, upon such
person’s promise to repay us therefore if it is ultimately determined that any such person shall not have been entitled to indemnification.
We
may undertake one or more significant corporate transactions that may not achieve their intended results, may adversely affect our financial
condition and our results of operations or result in unforeseeable risks to our business.
We
continuously evaluate the acquisition or disposition of operating businesses and assets and may in the future undertake one or more significant
transactions. Any such transaction could be material to our business and could take any number of forms, including mergers, joint ventures
and the purchase of equity interests. The consideration for such acquisitive transactions may include, among other things, cash, common
stock or equity interests in us or our subsidiaries, or a contribution of equipment to obtain equity interests, and in conjunction with
a transaction we might incur additional indebtedness. We also routinely evaluate the benefits of disposing of certain of our assets.
These
transactions may present significant risks such as insufficient revenue to offset liabilities assumed, potential loss of significant
revenue and income streams, increased or unexpected expenses, inadequate return of capital, regulatory or compliance issues, the triggering
of certain covenants in our debt agreements (including accelerated repayment) and unidentified issues not discovered in due diligence.
In addition, such transactions could distract management from current operations. As a result of the risks inherent in such transactions,
we cannot guarantee that any such transaction will ultimately result in the realization of its anticipated benefits or that it will not
have a material adverse effect on our business, financial condition and results of operations. If we were to complete such an acquisition,
disposition, investment or other strategic transaction, we may require additional debt or equity financing that could result in a significant
increase in our amount of debt and our debt service obligations or the number of outstanding shares of our common stock, thereby diluting
holders of our common stock outstanding prior to such acquisition.
The JOBS Act permits “emerging growth
companies” like us to take advantage of certain exemptions from various reporting requirements applicable to other public companies
that are not emerging growth companies.
We currently qualify as an “emerging growth
company” as defined in Section 2(a)(19) of the Securities Act, as modified by the JOBS Act. As such, we take and will continue to
take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth
companies for as long as we continue to be an emerging growth company, including: (i) the exemption from the auditor attestation requirements
with respect to internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act; (ii) the exemptions from say-on-pay,
say-on-frequency and say-on-golden parachute voting requirements; and (iii) reduced disclosure obligations regarding executive compensation
in our periodic reports and proxy statement/prospectus. As a result, our stockholders may not have access to certain information they
deem important. We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year: (a) following January
20, 2026, the fifth anniversary of our IPO; (b) in which we have total annual gross revenue of at least $1.07 billion; or (c) in which
we are deemed to be a large accelerated filer, which means the market value of our Common Stock that is held by non-affiliates exceeds
$700 million as of the last business day of our prior second fiscal quarter, and (ii) the date on which we have issued more than $1.0
billion in non-convertible debt during the prior three-year period.
In addition, Section 107 of the JOBS Act also
provides that an emerging growth company can take advantage of the exemption from complying with new or revised accounting standards provided
in Section 7(a)(2)(B) of the Securities Act as long as we are an emerging growth company. An emerging growth company can therefore delay
the adoption of certain accounting standards until those standards would otherwise apply to private companies. The JOBS Act provides that
a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies,
but any such election to opt out is irrevocable. We have elected to avail ourselves of such extended transition period, which means that
when a standard is issued or revised and it has different application dates for public or private companies, we, as an emerging growth
company, can adopt the new or revised standard at the time private companies adopt the new or revised standard. This may make comparison
of our financial statements with another public company that is neither an emerging growth company nor an emerging growth company that
has opted out of using the extended transition period difficult or impossible because of the potential differences in accounting standards
used.
We cannot predict if investors will find our Common
Stock less attractive because we rely on these exemptions. If some investors find our Common Stock less attractive as a result, there
may be a less active trading market for our Common Stock and our stock price may be more volatile.
We
have not paid dividends to date and do not intend to pay any dividends in the near future.
We
have never paid dividends on our common shares and presently intend to retain any future earnings to finance the operations of our business.
You may never receive any dividends on our shares.
If
large amounts of our common shares held by our existing stockholders are sold in the future, the market price of our common shares could
decline.
The
market price of our common shares could fall substantially if our existing stockholder were to sell large amounts of our common shares
in the public market following this offering. These sales, or the possibility that these sales may occur, could also make it more difficult
for us to sell equity or equity-related securities if we need to do so in the future to address then-existing financing needs. U.S. federal
securities laws requiring the registration or exemption from registration in connection with the sale of securities limit the number
of shares of common stock available for sale in the public market.
Investors
may suffer substantial dilution or an unrealized loss of seniority in preferences and privileges if we need to seek additional funding
in the future.
We
have authorized one billion shares of common stock with 21,263,515 shares outstanding. If we desire to raise additional capital in the
future to expand our operations, we may have to issue additional equity, preferred securities or convertible debt securities, which may
not need the approval of current shareholders. The issuance of new common shares would cause the buyers in this offering to suffer dilution
of their ownership percentage. In addition, it is possible that any future securities could grant new shareholders rights, preferences,
and/or privileges that are different from this offering.
We
may need additional capital in the future, which may not be available to us on favorable terms, or at all, and may dilute your ownership
of our common stock.
We
currently rely on outside financing and cash from operations to fund our operations, capital expenditures and expansion. We may require
additional capital from equity or debt financing in the future to:
| ● | respond
to competitive pressures; |
| ● | take
advantage of strategic opportunities, including more rapid expansion of our business or the
acquisition of complementary products, technologies or businesses; and |
| ● | develop
new products or enhancements to existing products. |
We
may not be able to secure timely additional financing on favorable terms, or at all. The terms of any additional financing may place
limits on our financial and operating flexibility. If we raise additional funds through issuances of equity, convertible debt securities
or other securities convertible into equity, our existing stockholders could suffer significant dilution in their percentage ownership
of the Company, and any new securities we issue could have rights, preferences and privileges senior to those of our common stock. If
we are unable to obtain adequate financing or financing on terms satisfactory to us, if and when we require it, our ability to grow or
support our business and to respond to business challenges could be significantly limited.
We
may expand through acquisitions of, or investments in, other companies or through business relationships, all of which may result in
additional dilution to our stockholders and consumption of resources that are necessary to sustain our business.
One
of our business strategies is to acquire competing or complementary services, technologies or businesses. In connection with one
or more of those transactions, we may:
| ● | issue
additional equity securities that would dilute our stockholders; |
| ● | use
cash that we may need in the future to operate our business; |
| ● | incur
debt on terms unfavorable to us or that we are unable to repay; |
| ● | incur
large charges or substantial liabilities; |
| ● | encounter
difficulties retaining key employees of the acquired company or integrating diverse business
cultures; |
| ● | become
subject to adverse tax consequences, substantial depreciation or deferred compensation charges;
and |
| ● | encounter
unfavorable reactions from investment banking market analysts who disapprove of our completed
acquisitions. |
Resales
of shares purchased by the Selling Stockholders may cause the market price of our common stock to decline.
We
are registering 10,027,925 shares of common stock which have been or are to be issued to the Selling Stockholders and 11,500,000 shares
of common stock issuable upon the exercise of our Public Warrants. The Selling Stockholders may also have the financial incentive to
sell their shares of common stock issuable to them in advance of or upon receiving such shares and to realize the profit equal to the
difference between the discounted price and the current market price of the shares. This may cause the market price of our common stock
to decline. The Selling Stockholders may offer and resell the common stock at a price and time determined by it, and the timing of sales
and the price at which the shares are sold could have an adverse effect upon the public market for our common stock. There may be no
independent or third-party underwriter involved in the offering of the shares held by or to be received by the Selling Stockholders,
and there can be no guarantee that the disposition of those shares will be completed in a manner that is not disruptive to the market
for our common shares.
If
we fail to comply with the continued listing requirements of Nasdaq, specifically, Nasdaq Listing Rules 5450(b)(2)(C) and 5450(b)(2)(A),
we may face possible delisting, which would result in a limited public market for our shares and make obtaining future debt or equity
financing more difficult for us.
As
previously reported, on August 16, 2024, the Company received a two letters from the Listing Qualifications Staff (the “Staff”)
of The Nasdaq Stock Market LLC (“Nasdaq”) notifying the Company that failed to maintain a minimum market value of publicly
held shares of at least $15,000,000 for a minimum of 10 consecutive business days and that the Company that failed to maintain a minimum
market value of listed securities must close of at lease $50,000,000 for a minimum of 10 consecutive business days. If the Company fails
to timely regain compliance with Nasdaq Listing Rules, the Company’s common stock will be subject to delisting from Nasdaq.
IN
ADDITION TO THE ABOVE RISKS, BUSINESSES ARE OFTEN SUBJECT TO RISKS NOT FORESEEN OR FULLY APPRECIATED BY MANAGEMENT. IN REVIEWING THIS
FILING, POTENTIAL INVESTORS SHOULD KEEP IN MIND THAT OTHER POSSIBLE RISKS MAY ADVERSELY IMPACT THE COMPANY’S BUSINESS OPERATIONS
AND THE VALUE OF THE COMPANY’S SECURITIES.
SPECIAL
NOTE REGARDING FORWARD LOOKING STATEMENTS
The
information herein contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and
Section 21E of the Securities Exchange Act of 1934, as amended. Actual results may materially differ from those projected in the forward-looking
statements as a result of certain risks and uncertainties set forth in this report. Although management believes that the assumptions
made and expectations reflected in the forward-looking statements are reasonable, there is no assurance that the underlying assumptions
will, in fact, prove to be correct or that actual results will not be different from expectations expressed in this report.
We
desire to take advantage of the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995. This filing
contains a number of forward-looking statements that reflect management’s current views and expectations with respect to our business,
strategies, products, future results and events, and financial performance. All statements made in this filing other than statements
of historical fact, including statements addressing operating performance, clinical developments which management expects or anticipates
will or may occur in the future, including statements related to our technology, market expectations, future revenues, financing alternatives,
statements expressing general optimism about future operating results, and non-historical information, are forward looking statements.
In particular, the words “believe,” “expect,” “intend,” “anticipate,” “estimate,”
“may,” variations of such words, and similar expressions identify forward-looking statements, but are not the exclusive means
of identifying such statements, and their absence does not mean that the statement is not forward-looking. These forward-looking statements
are subject to certain risks and uncertainties, including those discussed below. Our actual results, performance or achievements could
differ materially from historical results as well as those expressed in, anticipated, or implied by these forward-looking statements.
We do not undertake any obligation to revise these forward-looking statements to reflect any future events or circumstances.
Readers
should not place undue reliance on these forward-looking statements, which are based on management’s current expectations and projections
about future events, are not guarantees of future performance, are subject to risks, uncertainties and assumptions (including those described
below), and apply only as of the date of this filing. Our actual results, performance or achievements could differ materially from the
results expressed in, or implied by, these forward-looking statements. Factors which could cause or contribute to such differences include,
but are not limited to, the risks to be discussed in this Form S-1 Registration and in the press releases and other communications to
shareholders issued by us from time to time which attempt to advise interested parties of the risks and factors which may affect our
business. We undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information,
future events, or otherwise.
The
statements contained in this prospectus that are not historical fact are forward-looking statements which can be identified by the use
of forward-looking terminology such as “believes,” “expects,” “may,” “should,” or “anticipates”
or the negative thereof or other variations thereon or comparable terminology, or by discussions of strategy that involve risks and uncertainties.
We have made the forward-looking statements with management’s best estimates prepared in good faith.
Because
of the number and range of the assumptions underlying our projections and forward-looking statements, many of which are subject to significant
uncertainties and contingencies that are beyond our reasonable control, some of the assumptions inevitably will not materialize and unanticipated
events and circumstances may occur subsequent to the date of this prospectus.
These
forward-looking statements are based on current expectations, and we will not update this information other than required by law. Therefore,
the actual experience of the Company, and results achieved during the period covered by any particular projections and other forward-looking
statements should not be regarded as a representation by the Company, or any other person, that we will realize these estimates and projections,
and actual results may vary materially. We cannot assure you that any of these expectations will be realized or that any of the
forward-looking statements contained herein will prove to be accurate.
USE
OF PROCEEDS
We
will not receive any proceeds from the sale of the Shares by the Selling Stockholders. All net proceeds from the sale of the Shares covered
by this prospectus will go to the Selling Stockholders. We expect that the Selling Stockholders will sell their Shares as described under
“Plan of Distribution.”
We
may receive proceeds from the exercise of the Public Warrants and the Private Warrants to the extent that any Warrant are exercised for
cash. The Private Warrants, however, are exercisable on a cashless basis under certain circumstances. If all of the Warrants mentioned
above were exercised for cash in full, the proceeds would be approximately $138 million. We intend to use the net proceeds of such warrant
exercise, if any, for general corporate purposes, including working capital.
The
Selling Stockholders will pay any underwriting discounts and commissions and expenses incurred by the Selling Stockholders for brokerage,
accounting, tax or legal services or any other expenses incurred by the Selling Stockholders in disposing of the shares. We will bear
all other costs, fees and expenses incurred in effecting the registration of the shares covered by this prospectus, including, without
limitation, all registration and filing fees, fees and expenses of our counsel, certain expenses of counsel to the Selling Stockholders
and our independent registered public accountants.
PLAN
OF DISTRIBUTION
The
Selling Stockholders may, from time to time, sell any or all of shares of our Common Stock covered hereby on the NASDAQ Global Market,
or any other stock exchange, market or trading facility on which the shares are traded or in private transactions. The Selling Stockholders
may sell all or a portion of the shares being offered pursuant to this prospectus at fixed prices, at prevailing market prices at the
time of sale, at varying prices or at negotiated prices. The Selling Stockholders may use any one or more of the following methods when
selling shares:
| ● | ordinary
brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| ● | block
trades in which the broker-dealer will attempt to sell the shares as agent but may position
and resell a portion of the block as principal to facilitate the transaction; |
| ● | purchases
by a broker-dealer as principal and resale by the broker-dealer for its account; |
| ● | an
exchange distribution in accordance with the rules of the applicable exchange; |
| ● | privately
negotiated transactions; |
| ● | in
transactions through broker-dealers that agree with the selling stockholder to sell a specified
number of such shares at a stipulated price per share; |
| ● | through
the writing or settlement of options or other hedging transactions, whether through an options
exchange or otherwise; |
| ● | a
combination of any such methods of sale; or |
| ● | any
other method permitted pursuant to applicable law. |
The
Selling Stockholders may also sell shares under Rule 144 under the Securities Act, if available, rather than under this prospectus.
Broker-dealers
engaged by the Selling Stockholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions
or discounts from the selling stockholder (or, if any broker-dealer acts as agent for the purchaser of securities, from the purchaser)
in amounts to be negotiated, provided such amounts are in compliance with FINRA Rule 2121. Discounts, concessions, commissions and similar
selling expenses, if any, that can be attributed to the sale of Common Stock will be paid by the Selling Stockholders and/or the purchasers.
Any
broker-dealers or agents that are involved in selling the shares may be deemed to be “underwriters” within the meaning of
the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit
on the resale of the shares purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act, and
such broker-dealers or agents will be subject to the prospectus delivery requirements of the Securities Act.
Under
applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the resale securities may not simultaneously
engage in market making activities with respect to the Common Stock for the applicable restricted period, as defined in Regulation M,
prior to the commencement of the distribution. In addition, the selling stockholder will be subject to applicable provisions of the Exchange
Act and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of shares of
the Common Stock by the selling stockholder or any other person. We will make copies of this prospectus available to the selling stockholder
and have informed them of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale.
Under
the securities laws of some states, the common shares may be sold in such states only through registered or licensed brokers or dealers.
In addition, in some states the common shares may not be sold unless such shares have been registered or qualified for sale in such state
or an exemption from registration or qualification is available and is complied with.
The
Selling Stockholders will act independently of us in making decisions with respect to the timing, manner and size of each sale. There
can be no assurance that the selling stockholder will sell any or all of the shares of Common Stock registered pursuant to the registration
statement, of which this prospectus forms a part.
At
any time a particular offer of the shares of our Common Stock is made by the Selling Stockholders, a revised prospectus or prospectus
supplement, if required, will be distributed. Such prospectus supplement or post-effective amendment will be filed with the SEC to reflect
the disclosure of any required additional information with respect to the distribution of the shares of Common Stock. We may suspend
the sale of shares by the Selling Stockholders pursuant to this prospectus for certain periods of time for certain reasons, including
if the prospectus is required to be supplemented or amended to include additional material information.
Penny
Stock
Under
the rules of the Securities and Exchange Commission, our common stock will come within the definition of a “penny stock”
because the price of our common stock is below $5.00 per share. As a result, our common stock will be subject to the “penny
stock” rules and regulations. Broker-dealers who sell penny stocks to certain types of investors are required to comply with
the Commission’s regulations concerning the transfer of penny stock. These regulations require broker-dealers to:
Make
a suitability determination prior to selling penny stock to the purchaser;
| - | Receive
the purchaser’s written consent to the transaction; and |
| - | Provide
certain written disclosures to the purchaser. |
DESCRIPTION
OF BUSINESS
References
in this section to “we,” “our,” “us,” the “Company,” “ANEW,” or “ANEW
MEDICAL” generally refer to ANEW MEDICAL, INC., a Delaware corporation, and its consolidated subsidiaries.
ANEW
MEDICAL is dedicated to realizing the potential of biologic, cell and gene therapies to offer transformative patient outcomes in areas
of high unmet medical need by extending the reach of protein, cell, and gene therapies to highly prevalent neurodegenerative disorders
like amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease as they are universally fatal neurodegenerative diseases. Our
vision is to build a leading gene therapy company for the treatment of cancer and neurodegenerative diseases by progressing our α-Klotho
gene therapy research programs and identifying, developing, and commercializing other novel gene therapy treatments for neurodegenerative
diseases, cancer and other age-related pathologies.
We
have assembled a portfolio of protein and gene therapy candidates in partnership with leading scientific institutions and have built
a team with extensive experience in the biotechnology commercialization and gene therapy space. Our team will pursue new innovations
in vector design and delivery to optimize our investigational gene therapy product candidates for safety, potency, durability, and clinical
response. We plan on building integrated internal development capabilities from product development through commercialization and focus
on accelerating the pace of product development in the clinic. In addition, as part of our ongoing business strategy, we continue to
explore potential opportunities to acquire or license new product candidates as well as opportunities for partnership or collaboration
on our existing products in development.
There
are currently four technologies managed by our team — (1) A cell therapy and gene therapy platform that uses a gene
therapy approach to introduce a human gene sequence that produces a therapeutic protein called “Klotho” inside the body to
treat neurodegenerative diseases and other diseases of aging (a platform technology in-licensed from the Autonomous University of Barcelona
(UAB); (2) a proprietary, patented technology platform with a library of melanocortin receptor-binding molecules in-licensed from Teleost
Biopharmaceuticals LLC (Teleost) and the University of Arizona (UofA). The Company also acquired a (3) portfolio of “generic”
drugs and (4) a license to “biosimilar” biologics technology (in-licensed from RLS) that will allow us to sell hard-to-source,
difficult to find generic oncology drugs and off-patent biologic therapies. Our initial focus will be on our gene therapy research and
development programs. With an initial focus on the therapeutic potential of the human α-Klotho gene, we find that there is limited
competition investigating this target due to our intellectual property position relating to the secreted form of the Klotho protein (“s-KL”)
and technology know-how.
Our
Research Pipeline
ANEW
seeks to develop essential medicines for the treatment of chronic diseases — cancer, cardiovascular, muscle, skin, and neurodegenerative
disorders. Our cell and gene therapy platform consists of proprietary technology programs (patents issued and pending) that include (1)
a gene therapy program that uses a gene therapy approach to produce a therapeutic protein called “Klotho” inside the body
to treat neurodegenerative diseases and other diseases of aging (in-licensed pending patent applications from UAB), and (2) a library
of melanocortin receptor-binding molecules that activate or deactivate melanocortin receptors in the body that will be developed for
weight loss and eating disorders (in-licensed from Teleost and UofA). Next, our clinical-stage generic/biosimilar technologies consist
of (3) “off-patent” portfolio of generic drugs (4) and “off-patent” biosimilar biologics (in-licensed from RLS).
The
Company may develop all technologies, or it may decide to sell or partner and out-license certain technologies with other companies.
The
gene therapy product candidates are in the pre-clinical stage of development. The Company plans to seek market approval in countries
where we have issued and/or pending patents, to include the U.S., Canada, Europe, China and other viable markets.
ANEW
has conducted two pre-IND meetings with FDA regarding its biosimilars program. In the briefing package and at the meetings, ANEW presented
data showing similarity to reference products as obtained by our licensor, Reliance Life Sciences (RLS), using reference product obtained
from Genentech in the U.S. and Roche Pharmaceuticals in Switzerland. FDA insisted that ANEW test the U.S. reference product made by Genentech
for rituximab and for bevacizumab, and this must be completed before FDA would review any additional materials including the IND submission.
RLS has agreed to complete the biosimilarity testing package for both rituximab and bevacizumab using the U.S. reference products, Rituxan
and Avastin. After the testing is completed in late-2024, ANEW intends to meet again with FDA to discuss the IND and clinical protocol
strategy. Clinical trials for one or both antibodies could commence in the 2025 timeframe. RLS will manufacture both antibodies for the
clinical trials sponsored by ANEW. The protocols submitted in the IND applications will identify the number of patients in each control
(reference standard) and treatment (ANEW/RLS) arm of the study, and the primary and secondary endpoints are the same as those utilized
by the innovator and the other biosimilar antibody products approved to date — comparing six month overall response rates in both
arms of the study is the primary endpoint and comparative safety and PK/PD are the secondary endpoints.
Our
primary focus for 2024 and 2025 is the advancement of a sustainable portfolio of cell and gene therapy product candidates for age-associated
neurologic diseases, both rare “orphan diseases” as well as diseases in larger patient populations. The following table describes
our α-Klotho product pipeline.
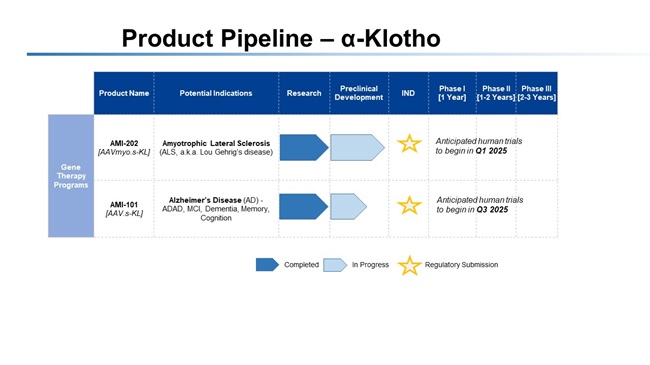
Cell
and Gene Therapy — α-Klotho gene
We
have licensed from the Autonomous University of Barcelona (UAB) the exclusive, worldwide rights to commercialize the α-Klotho sequence
called s-KL and know how for gene therapy and cell therapy against all neurodegenerative diseases — a longer-term, higher-risk
program, but high reward product development opportunities.
The
world’s population is aging rapidly and preserving brain health and body strength have emerged as major biomedical challenges.
Without novel interventions, over 80 million people worldwide will suffer from memory problems or physical disabilities resulting
from aging and age-related disease by 2040. Aging has been proven to be the primary risk factor for failing cognition and the development
of Alzheimer’s disease. Biologic or genetic regulators of aging might be harnessed for the treatment and prevention of cognitive
decline, depression, dementia, and sarcopenia (loss of muscle).
Because
aging is the primary risk factor for cognitive decline, and an emerging health threat to aging societies worldwide, providing medicine
like Klotho may, if proven, be able to counteract cognitive decline and neurodegenerative diseases such as Alzheimer’s disease.
Alzheimer’s disease (“AD”) is the most common type of senile dementia worldwide. Klotho is named after the Greek goddess
who “spins the thread of life”. A decade ago, Dr. Kuro-O et.al. established a transgenic knock-out mouse
without the Klotho gene. The mice were normal at birth but started to show multiple aging-related disorders such as arteriosclerosis,
osteoporosis, skin atrophy, loss of cognition, hyperphosphatemia, pulmonary emphysema, ectopic calcifications in the lung, heart and
skin, depletion of Purkinje cells in the brain, and infertility after 3-4 weeks—the α–kl+ mouse died at around
2 months whereas the normal mouse lives to around 2-3 years.
The
Klotho proteins produced by the Klotho gene is reported to significantly decline with age, especially in the brains of patients
with Alzheimer’s and animals genetically engineered to have AD. The in vitro treatment with Klotho protein of neurons taken
from mouse spinal cords can prevent neuronal death from toxic glutamate and beta-amyloid protein, while other studies have shown clearance
of beta-amyloid plaques in animals treated with the Klotho protein, so the utility of Klotho and s-KL in the treatment and prevention
of AD must be explored.
Therapeutic
approaches at increasing Klotho protein levels might theoretically prevent neuronal degeneration if treatment is started at the beginning
of disease and advance the outcome for AD patients. Recently, our investigator discovered a membrane-bound and a circulating secreted
form (“secreted-Klotho” or “s-KL”) of this protein hormone that is naturally found in humans and encoded by the
Klotho gene at a human chromosome locus of 13q12. We intend on investigating whether this hormone can play a role in protecting brain
neurons from degeneration, clearing beta-amyloid plaques, controlling the insulin/insulin-like growth factor signaling pathway, delaying
osteoporosis, lowering the incidence of cardiovascular disease, affecting kidney disease, and generally increasing the life-span of humans
and other mammals who have the gene.
The
upregulation of cerebral Klotho expression using a gene therapy approach has been evaluated by several investigators including
our Chief Scientific Advisor and inventor, Dr. Miguel Chillon Rodriguez and by Dr. Jun-Rong Du. In pioneering work, Chillon
Rodriguez and Masso et.al. (“α-Klotho Isoforms Have Different Spatio-Temporal Profiles in the Brain during Aging and Alzheimer’s
Disease”, PLOS ONE OI:10.1371/journal.pone.0143623 November 24, 2015) demonstrated an alternative RNA splicing variant of
the Klotho gene which produced a stable, truncated isoform of the hormone (“s-KL”) which, after intracranial, intrathecal,
and intravenous administration, can be detected directly as protein and mRNA in mouse protein tissue extracts. This work also showed
a strong correlation (p-value of 0.001) between high expression levels of the two Klotho transcripts, full-length, membrane-bound protein
(“m-KL”) and s-KL, in brain and healthy cognitive status while aging. Significantly, the secreted s-KL isoform is almost
exclusively (over 90%) found in brain, while m-KL is mostly expressed in kidney and to a lesser extent in brain. This suggests s-KL may
have an important role in the brain and overall brain health. More detailed studies revealed that the s-KL protein could be detected
in different murine brain regions involved in learning and memory processes, such as prefrontal cortex, motor cortex and hippocampus.
They found that the Klotho gene product, particularly the s-KL isoform, is a neuroprotective protein in mice and we believe that the
loss of this gene product leads to the onset and/or progression of cognitive deficits associated with aging. By modifying and increasing
s-KL levels in the brains of adult C57Bl/6J male mice, the AAV9 gene therapy vectors deliver the Klotho gene construct and the expression
of the Klotho protein is increased and cognition is improved. AAV9 vector is a recombinant adeno-associated virus serotype 9 delivery
vector for optimized delivery (“tropism”) to the central nervous system (CNS) tissues. Dr. Chillon Rodriguez will serve as
Co-Chair of the Combined Company’s Scientific Advisory Committee.
Chillon
Rodriguez and others have concluded that naturally occurring Klotho is a gene regulator of aging, increasing life expectancy when Klotho
is overexpressed by the gene, and that inhibition of the gene can accelerate the development of aging phenotypes. In mice, expression
levels of the secreted isoform Klotho (s-KL) are very high in the brain, suggesting that s-KL activity may have an important role in
the nervous system. The functional relevance at the behavioral level of modifying s-KL levels in the aging brain was studied using the
AAVrh10 and AAVrh9 vectors with tropism to the CNS to deliver and sustain expression of Klotho s-KL protein in 6- and 12-month-old normal
C57BL/6J mice. Study results demonstrated in animals that 6 months after a single injection of s-KL into the central nervous system,
long-lasting and quantifiable enhancement of learning and memory capabilities are found. More importantly, cognitive improvement was
observed in very elderly, 18-month-old mice treated once, at 12 months of age. These findings support the therapeutic potential
of s-KL as a treatment for cognitive decline associated with aging.
Dr. J-R
Du and associates have also successfully tested the utility of Klotho gene replacement in treatment and prevention of cognitive disorders
in mice. As we and many others have studied one theory of Alzheimer’s disease pathology, Alzheimer’s disease is characterized
by the presence of amyloid-beta (“A-β”) plaques. Du et.al. previously reported that Klotho lowered A-β levels in
the brain and protected against cognitive deficits in amyloid precursor protein/presenilin 1(APP/PS1) transgenic mice models. They induced
intracerebral Klotho overexpression in 13-month-old APP/ PS1 mice by injecting lentivirus that carried full-length mouse Klotho cDNA
in the lateral ventricle of the brain. They examined the effects of Klotho protein overexpression in the brain on cognition, A-β
burden, A-β-related neuropathology, microglia transformation, and A-β transport systems in vivo in this animal model. Additionally,
they investigated the effects of Klotho protein on A-β transport at the blood — cerebrospinal fluid barrier by knocking
down (inhibiting) Klotho production in primary human choroid plexus epithelial cells (HCPEpiCs).
Dr. Du
found that upregulation of Klotho levels in the brain and serum significantly ameliorated A-β burden, neuronal and synaptic loss
and cognitive deficits in aged APP/PS1 “AD mice”. Klotho gene therapy treatment significantly inhibited NACHT, LRR, and PYD
domain-containing protein 3 (NLRP3) and the subsequent transformation of microglia to the M2 type that may enhance microglia-mediated
A-β clearance. Meanwhile, Klotho overexpression also regulated A-β transporter expression, which may promote A-β transporter-mediated
A-β clearance. Moreover, the ability of human HCPEpiCs to transport A-β in vitro was also significantly impaired by
Klotho gene and protein knockdown. Given the observed neuroprotective effect of Klotho overexpression, the present findings provide convincing
evidence that Klotho gene and cell therapy should be further investigated as a potential therapeutic for Alzheimer’s disease and
other neurologic diseases.
Amyotrophic
lateral sclerosis (ALS) is a devastating neurodegenerative disorder of the motor system characterized by the degeneration of motoneurons
(MNs) and muscle denervation. This results in progressive muscle weakness ultimately leading to paralysis and patient death within 2-5
years of diagnosis, often due to respiratory failure1. Sporadic ALS (sALS) accounts for the majority of the cases, with only 5-10% being
familiar forms (fALS) caused by inherited mutations. Over 30 genes have been linked to ALS and several animal models carrying mutations
in these genes have been developed. The transgenic mouse featuring a high copy number of the human SOD1 gene with a glycine-to-alanine
transition at the 93rd codon referred to as SOD1G93A being the most extensively used.
Multiple
pathological mechanisms underlying the ALS disease process have been identified using ALS animal models. These mechanisms include oxidative
stress, inflammation, excitotoxicity, mitochondrial dysfunction, impaired proteostasis, endoplasmic reticulum stress, and RNA disturbances.
The interplay of these factors likely results in the selective death of motor neurons (MNs). Promising preclinical and clinical trials
targeting a single or a limited number of disease mechanisms have largely failed. A growing viewpoint supports a more integrated approach
by targeting multiple mechanisms simultaneously. ANEW proposes the therapeutic exploration of the pleiotropic RNA splice variant called
secreted protein α-Klotho (s-KL), which counteracts several mechanisms involved in neurodegeneration, such as oxidative stress,
demyelination, senescence, inflammation, and synaptic dysfunction. KL and s-KL are also critical for promoting muscle regeneration and
avoiding fibrosis.
Results
published recently by the Barcelona group have been promising (reference Bosch et.al., Research Square, titled “Muscle-targeted
Klotho Gene Therapy Ameliorates ALS Hallmarks by Addressing Multiple Disease Mechanisms in SOD1G93A Mice”, DOI: https://org/10.21203/rs.3.rs-4510655/V1),
Thus, the antiaging protein variant of α-Klotho (s-KL) exhibits neuroprotective and myoregenerative properties, mitigating age-related
neurodegeneration and promoting muscle regeneration in this mouse model of ALS. The study showed that s-KL harnesses its multifactorial,
pleiotropic properties in the context of Amyotrophic Lateral Sclerosis (ALS), a motoneuron disease lacking effective treatments due to
its diverse pathophysiological mechanisms. By overexpressing secreted KL in skeletal muscles of SOD1G93A mice with myotropic
viral vectors we aimed to directly protect muscles and exert a paracrine effect on motoneuron (MN) terminals. Secreted KL preserved MNs
and neuromuscular junctions, and mitigated glial reactivity, resulting in maintained muscle mass, improved neuromuscular function, delayed
disease onset, and extended survival. Even when administered during symptomatic stages, s-KL slowed down ALS progression. Transcriptomic
and proteomic studies in muscles revealed significant correction of pathophysiological mechanisms involved in ALS disease, unveiling
novel roles for KL. These findings highlight the potential application of muscle-secreted KL in ALS regardless of its origin and suggest
broader therapeutic implications.
We
are working with UAB scientists to advance our programs focused on neurodegenerative and age-related disorders — Alzheimer’s
and ALS — all based on the Klotho gene and the s-KL protein. In addition to our product pipeline candidates, we are building
a platform of next-generation gene and cell delivery technologies, to optimize our AAV-based, Lentivirus-based, and lipid-based gene
therapies. Much of this work will be funded by the Sponsored Research Agreement with UAB in Barcelona.
Supplementation
of this human Klotho protein by biologic infusion or by gene therapy may potentially treat many diseases and pathologies. Alzheimer’s
disease is extremely important, and certainly an urgent medical need, however the Klotho protein may be effective in heart disease, blood
vessel diseases, kidney diseases, and other diseases or problems associated with aging. Our plans are focused on these important programs,
but new programs may evolve from this core technology, such as gene-based therapy delivering other proteins of interest. We are developing
a gene therapy approach to introduce genetic material like plasmid DNA or mRNA into the body in order to induce production of a therapeutic
protein inside the patient’s body — we envision either blocking factors causing the disease, replacing human genes
that have shut down protein production, or replacing genes that have mutated to produce aberrant proteins that do not function at all.
Through gene therapy, or gene-modified stem cells, we aim to fix these problems.
In
gene therapy, the measurement of the concentration of the gene-transcribed protein is important. Thus, it is important to measure the
decrease or increase in blood and tissue levels of the Klotho gene-transcribed protein isoforms as part of ANEW’s gene therapy
development program. Of particular importance in our gene therapy program is the separation and measurement of the three Klotho protein
isoforms produced by the Klotho gene and the two Klotho protein metabolites. The human Klotho-gene encodes a type I transmembrane
protein (1012 amino acids, 140 kDa) with a large extracellular domain and a short intracellular portion (10 amino acids), called
“membrane Klotho” or m-KL predominantly expressed in the renal tubules and in the parathyroid glands. The extracellular
domain of Klotho cleaved from the m-KL and is found as a circulating factor called “plasma Klotho” or p-KL (1002 amino
acids, 135 kDa) that circulates and is detectable in blood and in lesser extent in other biological fluids. The p-KL is further metabolized
to two nearly-identical monomers (450 amino acids each, 70 kDa) called KL1 and KL2. The Klotho gene also produces
a RNA splice variant (550 amino acids, 63 kDa) called “secreted-Klotho” or s-KL. The s-KL isoform is predominantly
produced by cells in the brain and CNS.
Studies
addressing Klotho tissue concentrations used a commercial test kit, an antibody-based ELISA assay sold by IBL Laboratories (IBL Japan
and IBL-America), however the ELISA assay currently available is unable to differentiate the three isoforms, s-KL, m-KL, p-KL, and the
KL1 and KL2 metabolites (reference L. Pedersen et al. / Clinical Biochemistry 46 (2013) 1079 – 1083). The antibody
used in IBL’s commercial ELISA assay recognizes the same epitope sequence in the amino-terminal region of all the protein isoforms,
however the key differences of 13 amino acids are found in the carboxy-terminal end of the proteins. This difference is especially
true of the s-KL isoform compared to the p-KL and KL1 domains, which differ by a 13 amino acid “tail” found on s-KL. The
lack of specificity of the current commercial assays has motivated ANEW to develop a new assay method.
Therefore,
as part of the R&D program, ANEW plans to develop and validate an assay to separate and individually measure all Klotho isoforms
and metabolites of p-KL. We will compare ANEW’s assay to the IBL ELISA assay and establish assay specific reference intervals
in healthy and aged adults. In addition, several other clinical and laboratory characteristics will be determined in the cohorts and
compared to the levels of the circulating Klotho isoforms. The Klotho isoform assay will be used to identify subjects with low total
Klotho and evaluate the contribution of each Klotho isoform in the disease status of each subject. The specific measure of the s-KL isoform
produced as a result of the Klotho gene therapy product candidate will be analyzed and reported. If the assay developed as a research
test method (e.g. ELISA, SDS/Western blot, capillary electrophoresis) becomes a clinical biomarker of disease, ANEW will attempt to introduce
this test as a commercial diagnostic. ANEW scientists will develop isoform-specific monoclonal antibodies and utilize a separation technology
such as Western blot analysis, capillary electrophoresis or high performance liquid chromatography and mass spectrometry (LC/MS) to bring
this assay online in late 2024.
We
have in-licensed this key technology from UAB — with successful and encouraging data from animal models we hope
to study and treat these aging-related diseases. In some studies, the gene therapy that we are researching has shown evidence that it
may eliminate the disease-causing factors e.g. clearance of beta-amyloid plaques and “tau tangles” from the brain in AD or
prevention of mitochondrial oxidative stress and neuronal damage in ALS. We have not yet submitted any IND applications in connection
with any gene therapies.
Our
two lead gene therapy product candidates are AMI-101 (AAV9-CMV-sKL) for the treatment or prevention of Alzheimer’s disease, and
AMI-202 (AAVmyo-Des-sKL) gene therapy product for treatment and prevention of Lou Gehrig’s disease (amyotrophic lateral sclerosis
or “ALS”). Within the next 12 months, we plan to complete the animal toxicology package for AMI-202 and the submission of
an Investigational New Drug application (IND) to the FDA in 2025 for permission to start the first-in-human Phase I “Compassionate
Use” study of AMI-202 in late-stage ALS patients. Likewise, the pre-clinical development program for AMI-101, similar to that of
AMI-202, will follow behind the development of AMI-202 by six to nine months. We plan to find a corporate partner to help in the
development of AMI-101 for the Alzheimer’s disease indication. We may also bring the recombinant s-KL protein into clinical trials
to evaluate the safety and efficacy of infusing the protein into normal healthy volunteers for pharmacology (“PK/PD”) and
safety, and then in select patient populations. Our AMI-101 and AMI-202 product candidate are dependent upon the s-KL variant and the
intellectual property position around the RNA splice variant. We have in-licensed the exclusive rights to patents and patent applications
to treat neurodegenerative diseases with patent approvals in the U.S. (US Patent No. 12,036,268), Europe, China, Hong Kong, and pending
in Canada. Patent applications ( PCT Application No. PCT/EP2023/059677) filed in the major markets covering the composition and
use of s-KL gene therapies to treat neuromuscular diseases like ALS are scheduled for review by the examiners in these countries.
Also,
our team will pursue new innovations in vector design and delivery to optimize our investigational gene therapy products for safety,
potency, durability, and clinical response. We will continue to build integrated internal development capabilities from product development
through commercialization and focus on accelerating the pace of product development in the clinic. As part of our ongoing business strategy,
we continue to explore potential opportunities to acquire or license new product candidates as well as opportunities for partnership
or collaboration on our existing products in development.
Intellectual
Property — Our Licenses and Technology
We
have entered into other license agreements, some exclusive, covering the key technologies upon which our research and product development
efforts will be based. Details of the licenses are below.
Gene
and Cell Therapy Programs
UAB
Agreements:
1.
On January 20, 2022, we entered into an exclusive, worldwide, royalty-bearing license with Universitat Autònoma de Barcelona
(“UAB”) and Institució Catalana De Recerca I Estudis Avançats (“ICREA”) to develop and commercialize
certain patent rights, technology and know-how in the field of secreted-Klotho splicing variant diagnostics and therapeutics to prevent
or treat neurodegenerative diseases and other age-related pathologies of the central nervous system. The patents (issued in Europe and
China) and pending patent applications support our ANEW101 and ANEW202 product candidates with claims directed to methods of treatment
and compositions of matter. The issued patents and any applications granted on a pending application would be enforceable until 11/21/2036,
absent any patent term adjustment or extension. The patent family includes:
| Country |
|
Application/Publication |
|
Status |
| US |
|
12,036,268 |
|
Issued |
| EU |
|
EP3377091B1 |
|
Issued |
| Canada |
|
CA3005398A |
|
Pending |
| China |
|
CN108289933B |
|
Issued |
| Hong Kong |
|
HK1259628A1 |
|
Issued |
| Japan |
|
JP2019501643A |
|
Pending |
The
license includes the right to sublicense. The term of the license extends until the expiration of the last to expire Royalty Term, on
a product-by-product, country-by-country basis, unless terminated earlier pursuant to the terms of the agreement. “Royalty term”
means the period of time, product-by-product, country-by-country, beginning on the first commercial sale and extending to the later of
(i) last patent to expire such Product in such country, or (ii) indefinitely (if no patent right covers such Product in such
country), provided that no generic equivalent or biosimilar product is commercialized in such country. UAB and ICREA reserve rights to
use the licensed rights for internal non-commercial research and to allow other academic institutions to use the licensed rights for
internal non-commercial research.
As
set forth in the agreement, we are responsible for all costs associated with the research, development and commercialization of the Products,
including entering into a Sponsored Research agreement with Dr. Chillon’s and Dr. Bosch’s laboratory at UAB (discussed
below).
As
consideration for the license, we paid an upfront fee corresponding to prior patent costs. We are also required to pay an annual patent
maintenance fee of 10.000€ as well as certain milestone payments and a royalty of 3% of Net Sales, as that term is defined in the
agreement, in countries wherein patent rights exist, otherwise the royalty is 1.5% of Net Sales. Separate royalty amounts exist in the
event we sublicense any rights granted. The milestone payments are 35,000 euro upon IND achievement, 250,000 euro upon completion of
Phase 1 study, 500,000 euro upon completion of Phase 2 study, 1,200,000 euro upon completion of Phase 3 study and 2,000,000
euro upon first commercial product approval in the U.S., EU or Japan. We are also responsible for all ongoing patent maintenance, protection
and management costs.
The
parties may terminate the agreement in the event of a material default by the other party of any terms, conditions, obligations and undertakings
upon written notice which is not cured within three months from such notice. UAB/ ICREA may terminate if we (1) abandon or
discontinue use of the licensed rights for more than one year, (2) illegally use the licensed rights, including violations of the
Universal Declaration of Human Rights, (3) fail to make royalty payments or underreporting such payments in excess of 15% in two
different years; (4) challenge the validity or enforceability of the licensed patent rights; or (5) fail to achieve product
development stages set forth in the agreement. We may terminate the agreement at any time with 90 days’ notice.
2.
On December 20, 2022, we entered into an exclusive, worldwide royalty-bearing license with UAB ICREA, Consorcio Centro de Investigación
Biomédica en Red (“CIBER”), and Fundació Hospital Universitari Vall D’hebron — Institut
de Recerca (“VHIR”) to develop and commercialize certain patent rights, technology and know-how in the field of Neuronal
(central nervous system) or Neuromuscular (peripheral nervous system) Diseases or Disorders. The pending patent application supports
our ANEW202 product candidate with claims directed to methods of treatment and compositions of matter. If granted, such patent would
be enforceable until 4/16/2043, absent any patent term adjustment or extension. The patent family includes:
| Country |
|
Application
Number |
|
Filing
date |
|
Status |
| US |
|
63/330,684 |
|
April 26,
2022 |
|
Expired |
| US |
|
18/299,989 |
|
April 13,
2023 |
|
Pending |
| PCT |
|
PCT/EP2023/059677 |
|
April 13,
2023 |
|
Pending |
The
license includes the right to sublicense. The term of the license extends until the expiration of the last to expire Royalty Term, on
a product-by-product, country-by-country basis, unless terminated earlier pursuant to the terms of the agreement. “Royalty term”
means the period of time, product-by-product, country-by-country, beginning on the first commercial sale and extending to the later of
(i) last patent to expire such Product in such country, or (ii) indefinitely (if no patent right covers such Product in such
country), provided that no generic equivalent or biosimilar product is commercialized in such country. UAB/ICREA/CIBER/VHIR reserve rights
to use the licensed rights for internal non-commercial research and to allow other academic institutions to use the licensed rights for
internal non-commercial research.
As
set forth in the agreement, we are responsible for all costs associated with the research, development and commercialization of the Products,
including entering into a Sponsored Research agreement with Dr. Bosch and/or Dr. Chillon’s laboratory at UAB to perform
R&D activities (discussed below).
As
consideration for the license and in the event that our January 20, 2022 license discussed above is terminated, then we must pay an annual
patent maintenance fee of 10.000€. We will also be obligated to pay certain milestone payments and a royalty of 3% of Net Sales,
as that term is defined in the agreement, in countries wherein patent rights exist, otherwise the royalty is 1.5% of Net Sales. Separate
royalties exist in the event we sublicense any rights granted. The milestone payments are 35,000 euro upon IND achievement, 250,000 euro
upon completion of Phase 1 study, 500,000 euro upon completion of Phase 2 study, 1,200,000 euro upon completion of Phase 3
study and 2,000,000 euro upon first commercial product approval in the U.S., EU or Japan. The agreement has an anti-stacking clause in
the event that Products are covered by patents or technology from both this agreement and our 1/20/2022 license discussed above, then
we are only obligated to pay royalties of such Product pursuant to this agreement. We are also responsible for all ongoing patent maintenance,
protection and management costs.
The
parties may terminate the agreement in the event of a material default by the other party of any terms, conditions, obligations and undertakings
upon written notice which is not cured within three months from such notice. UAB/ICREA/CIBER/VHIR may terminate if we (1) abandon
or discontinue use of the licensed rights for more than one year, (2) illegally use the licensed rights, including violations of
the Universal Declaration of Human Rights, (3) fail to make royalty payments or underreporting such payments in excess of 15% in
two different years; (4) challenge the validity or enforceability of the licensed patent rights; or (5) fail to achieve
product development stages set forth in the agreement. We may terminate the agreement at any time with 90 days’ notice.
3.
As mentioned above, and in accordance with our UAB licenses, we entered into a Sponsored Research Agreement with UAB on January 24,
2022, under which the research will be directed and supervised by the Principal Investigators, Dr. Assumpcio Bosch and Dr. Miguel
Chillon having a two year budget of 623.100,00€.
University
of Heidelberg Agreement:
On
March 5, 2023, entered into a non-exclusive, worldwide license with University of Heidelberg, Germany for innovations related to
certain modified AAV capsid polypeptides in the field of neuromuscular disorders. The pending patent applications support our ANEW202
product candidate with claims directed to methods of treatment and compositions of matter. If granted, such patent would be enforceable
until 4/26/2039, absent any patent term adjustment or extension. The patent family includes:
| Country |
|
Application
Number |
|
Status |
| PCT |
|
PCT/EP2019/060790 |
|
Nationalized |
| CA |
|
3,097,375 |
|
Pending |
| US |
|
17/051,123 |
|
Pending |
| EP |
|
3784288 |
|
Pending |
| AU |
|
2019258830 |
|
Pending |
| JP |
|
2020-560127 |
|
Pending |
| CN |
|
201980028398 |
|
Pending |
The
license includes the right to sublicense. The term of the license extends until the expiration or cancelation of each patent in each
licensed jurisdiction.
As
consideration for the license, we are required to pay the licensor certain milestone payments and a royalty of 2% of Net Sales, as that
term is defined in the agreement. The milestone payments are 150,000 euro upon execution of the license, 150,000 euro within 60 days
of start of Phase 1 clinical trial, and 200,000 euro within 60 days of start of Phase 3 trial.
The
University of Heidelberg may terminate the agreement (1) in the event of a material default by us upon written notice which is not
cured within 60 days from such notice, (2) with 10 days’ notice if we cease operations, file for bankruptcy, dissolution
or make an assignment to creditors, or (3) if we challenge the licensed patents. We may terminate the agreement at any time with
120 days’ notice.
Melanocortin
Receptor-Binding Ligands Programs
Teleost
Biopharmaceuticals, LLC Agreement:
On
January 27, 2023, we entered into a Sublicense Agreement with Teleost Biopharmaceuticals, LLC (“Teleost”) and the University
of Arizona for the exclusive worldwide right to develop and commercialize certain patent rights owned by University Arizona Board Regents,
and licensed to Teleost, as well as an exclusive worldwide license to certain technology and know-how rights owned by Teleost in the
field of gamma-melanocortins comprised of drug and peptide chemicals, API and pharmaceuticals that bind, affect, and potentially treat
diseases directly or indirectly related to human melanocortin receptors that do not compete or interfere with Teleost’s clinical
cosmetic indications for use. The sublicensed patents arise from six families that claim compositions of matter and/or methods of use.
The patents pertain to product candidates for treatment of weight loss and eating disorders, dermal pathologies, for treatment of depression,
stress (PTSD), and potentially cognition:
1
— Melanocortin 1 Receptor Ligands And Methods Of Use PCT/US2012/038425 (WO 2012/158960)
| US
Patent or Application Number |
|
Expiration |
|
Status |
|
Foreign
Counterparts |
| 9,441,013 |
|
5/17/2032 |
|
Issued |
|
none |
| 10,329,326 |
|
5/17/2032 |
|
Issued |
|
|
| 11,230,568 |
|
5/17/2032 |
|
Issued |
|
|
| 17/582,368
(filed 1/24/2022) |
|
n/a |
|
Pending |
|
|
2
— Melanocortin Ligands For Skin Care
| US
Patent or Application Number |
|
Expiration |
|
Status |
|
Foreign
Counterparts |
| 9,290,539 |
|
5/17/2032 |
|
Issued |
|
none |
| 9,539,301 |
|
5/17/2032 |
|
Issued |
|
|
3
— Novel Modulators of Melanocortin receptors PCT/US2015/035180 (WO2015191765)
| US
Patent or Application Number |
|
Expiration |
|
Status |
|
Foreign
Counterparts |
| 11,124,542 |
|
5/17/2032 |
|
issued |
|
EU
3,177,737 |
4
— Enhanced melanoma cancer prevention by novel melanotropins PCT/US2016/033010 (WO2016187264)
| US
Patent or Application Number |
|
Expiration |
|
Status |
|
Foreign
Counterparts |
| 10,188,704 |
|
5/18/2036 |
|
issued |
|
CA2986423C
EP3297655B1 |
The
license includes the right to sublicense. The term of the license extends until the last to expire patents. We are required to pay the
licensor (1) a license fee of $60,000, (2) ongoing costs associated with the filing and maintenance of the patents, as well
as (3) certain milestone payments and a royalty of 10% of Net Sales, as that term is defined in the agreement. The milestone payments
are $ $50,000 at each IND approval and $15,000 at each CTA approval in Europe, $100,000 at the start of each Phase 3 trial, $250,000
at first market approval outside the US, $500,000 at FDA approval to market in US, $150,000 at first commercial approval of each product
outside the US, $500,000 at first commercial approval of each product with the US, and $1,000,000 when commercial sales of $100 million
or more of each licensed product. We are also obligated to pay a 15% royalty on all fees received from a sublicensee as well as patent
maintenance fees of $20,000 in 2025, $30,000 in 2026, $50,000 in 2027, $100,000 in 2028 and each year thereafter. Our license maintenance
fees are credited against our royalties due.
The
parties may terminate the agreement in the event of a material default by the other party of any terms, conditions, obligations and undertakings
upon written notice which is not cured within 30 days from such notice. In addition, either party may terminate if the other party
becomes bankrupt or insolvent. We may terminate the agreement at any time with 180 days’ notice in the event of a product
development failure or if we determine that the product is no longer commercially viable.
Biosimilars
Program
Reliance
Life Science Agreements:
On
November 27, 2014, we entered into an exclusive, royalty bearing License Agreement with Reliance Life Sciences Private Limited (“RLS”)
to develop, manufacture finished Product(s) based on an API for a finished biosimilar to Roche’s rituximab (MabThera®/Rituxan®),
trastuzumab (Herceptin®), and bevacizumab (Avastin®) in the Field of the treatment of cancer, autoimmune
diseases, neovascularization diseases, and other diseases or conditions that may be treated by the Products or derivatives (e.g. antibody-drug
conjugates) of the Products. The license permits our exclusive use in the Territory of RLS’s technology (including RLS’s
Know-how concerning manufacturing and RLS regulatory data, including RLS’s dossier for India). The licensed Territory is limited
to North America, Europe and Israel. Future patents related to the Product would be included in the agreement, however there were no
RLS patents in existence as of the execution of the agreement.
As
consideration we previously paid RLS upfront license fees of $300,000 and are required to pay the licensor certain milestone payments
and a royalty of 5% of Net Sales, as that term is defined in the agreement. The milestone payments are per Product of: $250,000 upon
submission to the FDA, $100,000 upon submission of Market Authorization to European Medicines Agency (“EMA”), $3,000,000
upon receipt of Marketing Authorization from US Food and Drugs Administration, $2,000,000 upon approval in Europe by EMA, $500,000 upon
market authorization in Canada, $300,000 upon market authorization in Mexico, $200,000 upon market authorization in Israel, $2,000,000
upon Net Sales of $20 million or more in US, $1,000,000 upon Net Sales of $10 million or more in Europe, and $10,000,000 upon
achieving Net Sales in any country of over $100 million. The license includes the right to sublicense. In the event we sublicense
rights under this agreement will subject us to alternate sublicense milestone fees.
The
term of the license is for a period of 10 years after Market Authorization on a country-by-country basis, which automatically extends
for another 10 years unless notice not to extend is received 180 days prior to the end of the term.
The
parties may also terminate the agreement at any time if (1) the other party breaches the agreement and fails to cure the breach
within 60 days of notice of such breach, (2) bankruptcy of the other party, or (3) a change in the law so that performance
by a party becomes onerous or inexpedient. We may also terminate this agreement on a product-by-product basis upon 60 days written
notice. In the event that we fail to make any payment, RLS may terminate the agreement by giving 30 days prior written notice.
Pursuant
to the license, RLS will exclusively provide to us the API pursuant to a separate manufacturing and supply agreement that the parties
executed on November 27, 2014.
On
November 27, 2014 the parties executed amendments to the above license and manufacturing and supply agreements to include development
and manufacture of cetuximab (Erbitux®, BMS/Lilly/Imclone) under the same terms, exclusive rights and territories. As
consideration for the additional rights set forth in the amendment, we paid RLS upfront license fees of $150,000 and are required to
pay the licensor certain milestone payments and a royalty of 5% of Net Sales, as that term is defined in the agreement. The milestone
payments are: $250,000 upon submission to the FDA, $100,000 upon submission of Market Authorization to EMA, $3,000,000 upon receipt of
Marketing Authorization from US Food and Drugs Administration, $2,000,000 upon approval in Europe by EMA, $500,000 upon market authorization
in Canada, $300,000 upon market authorization in Mexico, $200,000 upon market authorization in Israel, $2,000,000 upon Net Sales of $20 million
or more in US, $1,000,000 upon Net Sales of $10 million or more in Europe, and $10,000,000 upon achieving Net Sales in any country
of over $100 million. The license includes the right to sublicense. In the event we sublicense rights under this agreement will
subject us to alternate sublicense milestone fees.
Avastin®
and Rituxan®/Mabthera® are the brand names of bevacizumab and rituximab. Both Genentech in the U.S. and
Roche in Switzerland manufacture these antibodies. Rituximab is marketed as Rituxan® in the US and Japan, and branded
as Mabthera® outside the US and Japan.
The
approved indications for these antibodies are as follows:
Rituxan®
(rituximab) is indicated for the treatment of adult patients with Non-Hodgkin’s Lymphoma (NHL) — Relapsed or refractory,
low-grade or follicular, CD20-positive, B-cell NHL as a single agent, Previously untreated follicular, CD20-positive, B-cell NHL in combination
with first line chemotherapy and, in patients achieving a complete or partial response to a rituximab product in combination with chemotherapy,
as single-agent maintenance therapy, Non-progressing (including stable disease), low-grade, CD20-positive, B-cell NHL, as a single agent,
after first-line CVP chemotherapy, Previously untreated diffuse large B-cell, CD20-positive NHL in combination with CHOP or other anthracycline-based
chemotherapy regimens. Rituxan is indicated for the treatment of pediatric patients aged 6 months and older with mature B-cell NHL
and mature B-cell acute leukemia (B-AL), Previously untreated, advanced stage, CD20-positive, diffuse large B-cell lymphoma (DLBCL),
Burkitt lymphoma (BL), Burkitt-like lymphoma (BLL) or mature B-cell acute leukemia (B-AL) in combination with chemotherapy. Rituxan is
indicated for the treatment of adult patients with previously untreated and previously treated CD20-positive Chronic Lymphocytic Leukemia
(CLL) in combination with fludarabine and cyclophosphamide (FC).
Rituxan,
in combination with methotrexate, is also indicated for the treatment of adult patients with moderately to severely active rheumatoid
arthritis (RA) who have had an inadequate response to one or more TNF antagonist therapies. Rituxan is also indicated in combination
with glucocorticoids, is indicated for the treatment of adult and pediatric patients 2 years of age and older with Granulomatosis
with Polyangiitis (GPA) (Wegener’s Granulomatosis) and Microscopic Polyangiitis (MPA). Rituxan is indicated for the treatment of
adult patients with moderate to severe pemphigus vulgaris (PV).
Bevacizumab
(Avastin®) is indicated for the treatment of the following indications: Stage III or IV ovarian cancer (OC)
after primary surgery — Avastin, in combination with carboplatin and paclitaxel, followed by Avastin as a single agent, is indicated
for the treatment of patients with stage III or IV epithelial ovarian, fallopian tube, or primary peritoneal cancer following
initial surgical resection. Recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer (rOC) -Avastin, in combination
with paclitaxel, pegylated liposomal doxorubicin, or topotecan, is indicated for the treatment of patients with platinum-resistant recurrent
epithelial ovarian, fallopian tube or primary peritoneal cancer who received no more than 2 prior chemotherapy regimens. Avastin, in
combination with carboplatin and paclitaxel, or with carboplatin and gemcitabine, followed by Avastin as a single agent, is indicated
for the treatment of patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer. Persistent,
recurrent, or metastatic cervical cancer (CC) — Avastin, in combination with paclitaxel and cisplatin or paclitaxel and topotecan,
is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer.
Metastatic
renal cell carcinoma (mRCC) — Avastin, in combination with interferon alfa, is indicated for the treatment of metastatic renal
cell carcinoma.
Recurrent
glioblastoma (rGBM) — Avastin is indicated for the treatment of recurrent glioblastoma in adults.
First-line
non-squamous non-small cell lung cancer (NSCLC) — Avastin, in combination with carboplatin and paclitaxel, is indicated for the
first-line treatment of patients with unresectable, locally advanced, recurrent or metastatic non-squamous non-small cell lung cancer.
Metastatic
colorectal cancer (MCRC) — Avastin, in combination with intravenous fluorouracil-based chemotherapy, is indicated for the first-
or second-line treatment of patients with metastatic colorectal cancer. Avastin, in combination with fluoropyrimidine-irinotecan- or
fluoropyrimidine-oxaliplatin-based chemotherapy, is indicated for the second-line treatment of patients with metastatic colorectal cancer
who have progressed on a first-line bevacizumab product-containing regimen. Limitation of Use: Avastin is not indicated for adjuvant
treatment of colon cancer.
Hepatocellular
carcinoma (HCC) — Avastin, in combination with atezolizumab, is indicated for the treatment of patients with unresectable or metastatic
hepatocellular carcinoma who have not received prior systemic therapy.
Competition
There
are three classes of drugs that are used to help with the physiologic effects of advancing cognitive disorders and dementia, (1) Cholinesterase
inhibitors — donepezil (Aricept), rivastigmine (Elexon) and galantamine (Razadyne), (2) the Glutamate regulators — memantine
(Namenda) and (3) the combination of cholinesterase inhibitor and glutamate regulator called Namzaric) donepezil plus memantine._Companies
selling these generic and branded drugs include Allergan, Eisai Co., Ltd., Novartis AG, Daiichi Sankyo Company, Limited., Merz Pharma,
Pfizer Inc., Johnson & Johnson Services, Inc., H. Lundbeck A/S, Biogen, AstraZeneca, F. Hoffmann-La Roche Ltd, VTV
Therapeutics, TauRx, Eli Lilly and Company, Teva Pharmaceutical Industries Ltd., Ono Pharmaceutical Co., Ltd., AC Immune, AB Science,
AbbVie Inc., Bristol-Myers Squibb Company, Takeda Pharmaceutical Company Limited., Bayer AG, and among others.
The
anti-amyloid antibodies, aducanumab (Biogen:Aduhelm) and lecanumab (Lilly:Leqembi) were FDA approved and will potentially compete with
Roche, Bristol-Myers/Celgene, and others developing anti-amyloid antibodies. Recently, aducanumab (Biogen:Aduhelm) was removed from the
market due to lack of effectiveness and serious side effects. The long-term prognosis of anti-amyloid antibodies is still questionable
based on the failure of so many anti-amyloid product candidates. There remains multiple large and early stage pharma companies pursuing
therapeutics against Alzheimer’s disease. Very few companies mention the use of Klotho or Klotho gene therapy on their websites
and/or in press releases. ADvantage Therapeutics mentions Klotho in the website, but they appear to be working on AD04™ and suggest
it may act as an immunomodulator, stimulating and/or regulating the immune system to reduce AD pathology. There is mention of mRNA therapies,
but this vaccine adjuvant AD04 appears to be their main product candidate, and no description of their Klotho program. They may be testing
Klotho as an immunomodulator. Another company, Bioviva, says on their website that “Klotho has been shown to reverse beta- amyloid
plaques” but gives no details on their stage of product development. One of their team members, George Church, published an article
on full-length Klotho m-KL about 10 years ago, so it may be full-length Klotho. Bioviva don’t mention Klotho or Klotho product
stage of development at their website.
To
the best of our knowledge, there is no direct competition in ALS, AD, PD or other neurodegenerative disorders involving the human s-KL
Klotho gene or the s-KL protein. Two companies, Klotho Therapeutics and Klogenix, were formed many years ago but the management
teams could not raise capital and they are now defunct.
Biosimilar
Competition
ANEW
will face competition in the US, European and other markets from the innovator brand in that market, from biosimilars already approved
in those markets, and from biosimilars still in development in those markets. Major players in the rituximab biosimilars market are Pfizer
Inc., Mylan Inc., Amgen Inc., Teva Pharmaceutical Industries Ltd., and Celltrion Healthcare Co. Ltd., Sandoz International GmbH, Reliance
Life Sciences, C.H. Boehringer Sohn AG & Co. KG, BioXpress Therapeutics SA, and Intas Biopharmaceuticals Ltd. There are two other
Avastin biosimilars, Mvasi (bevacizumab-awwb), co-developed by Amgen and Allergan, and Pfizer’s Zirabev (bevacizumab-bvzr).
In
Europe, due to immediate acceptance by physicians and government agencies of potential savings due to the price competition of biosimilars,
the prices of the innovator’s products came down by 30%-40% or more because of the lower retail prices of the biosimilar product
once approved by EMA, whereas in the U.S., biosimilar products approved by FDA are priced at nearly the same price of the innovator’s
product. Now, with improving acceptance of biosimilar products in the U.S., prices of both the biosimilar and innovator’s products
are coming down, and significant savings are now being realized by the insurance companies and other payors in the U.S.
Cell
and Gene Therapy Competition — Configured for specific rare diseases
Gene
Therapy of Alzheimer’s
Researchers
at University of California San Diego School of Medicine have launched a first-in-human Phase I clinical trial (March 2021)
to assess the safety and efficacy of a gene therapy to deliver a key protein into the brains of persons with Alzheimer’s disease
(AD) or Mild Cognitive Impairment (MCI), a condition that often precedes full-blown dementia. The protein, called brain-derived neurotrophic
factor or BDNF, is part of a family of growth factors found in the brain and central nervous system that support the survival of existing
neurons and promote growth and differentiation of new neurons and synapses. BDNF is particularly important in brain regions susceptible
to degeneration in AD. In previous published research, principal investigator Mark Tuszynski, MD, PhD, professor of neuroscience
and director of the Translational Neuroscience Institute at UC San Diego School of Medicine has helped to bring this approach forward.
No one else to our knowledge is actively pursuing this approach for AD or ADAD.
The
leading competitor in Klotho gene therapy is Unity Biotechnology, a San Francisco Bay area startup biotechnology company that develops
drugs which target senescent cells. Their website claims the company’s products in development include UBX1325 that targets Bcl-xL,
a mechanism to eliminate senescent cells in age-related eye diseases, and UBX1967, a preclinical product targeting ophthalmologic diseases.
They claim to be in “lead optimization” of a Klotho product candidate, but they have not reported any data and no pre-clinical
or clinical trials data published to date. Unity recently licensed the Dubal Klotho technology to two of their publication authors who
formed Jocasta Therapeutics, Inc. and a hedge fund in Northbrook, IL, and no press releases have been published. They would be pursuing
full-length, m-KL or p-KL, and not s-KL.
Gene
Therapy and Other Approaches to ALS
Gene
therapy approaches to ALS are being pursued using anti-sense oligonucleotides to block ataxin-2 (Biogen), and to try to produce Caveolin-1
(Eikonoklastes Therapeutics), which are believed to be involved with innervation of muscles cells. Neurosense Therapeutics is conducting
a clinical trial of a combination of two pharmaceuticals, ciprofloxacin and celecoxib, an antibiotic and an anti-inflammatory, to try
to reduce inflammatory reactions in the muscles. Amylyx received approval by FDA of Relyvrio®, a combination of sodium
phenylbutyrate (an aromatic fatty acid salt used in urea cycle disorders) and taurursodiol (a bile acid used in treatment of gall stones
and several other diseases including ALS). Recently, Amylyx removed Relyvrio® from the market for lack of efficacy in
follow-on studies. None of these potential agents have shown significant clinical benefit and muscle-saving activity in human studies
to date.
Gene
Therapy for Parkinson’s Disease
Parkinson’s
disease is a progressive neurodegenerative disorder that is caused by the loss of dopamine signaling in the brain and results in the
continual decline of motor control and quality of life. While there is currently no cure for the disease, through dopaminergic strategies
that temporarily increase dopamine levels in the brain, patients see a short-term improvement in symptoms which wanes with disease progression.
AXO-Lenti-PD is the only investigational gene therapy for Parkinson’s disease that delivers the three key genes (TH, CH1, and AADC)
required for endogenous dopamine synthesis in a single lentiviral vector. The goal of this one-time infusion is to restore steady, tonic
levels of dopamine, potentially reducing the need for daily L-dopa medication while stabilizing the disease to provide long-lasting benefits.
Initial clinical data demonstrate that a single dose of AXO-Lenti-PD “turns back the clock” for patients by improving motor
function and activities of daily living. AXO-Lenti-PD has been optimized from ProSavin, an earlier gene therapy for the treatment of
Parkinson’s disease. The company is currently evaluating AXO-Lenti-PD in a Phase 2 clinical trial (SUNRISE-PD) in patients
with moderate to advanced Parkinson’s disease. These companies have declared bankruptcy and none are in business.
Government
Regulations
Regulations
by governmental authorities in the U.S. and other countries are a significant factor in developing manufacturing and marketing of our
pharmaceutical products. The nature and extent to which such regulation applies to us may vary depending on the nature of our product
candidates. We anticipate that all of our product candidates will require regulatory approval by governmental agencies prior to commercialization.
Our product candidates are subject to rigorous pre-clinical testing and clinical trials and other approval procedures of the FDA, and
similar regulatory authorities in Europe, Japan and other countries. Various governmental statutes and regulations also govern or influence
clinical trials, Chemistry, Manufacture and Control (CMC) related to such product candidates and their marketing. The approval process
and subsequent compliance with related statutes and regulations require substantial time and capital commitment, and there can be no
guarantee that approvals will be granted.
In
the United States, the FDA regulates pharmaceutical and biological products under the Federal Food, Drug, and Cosmetic Act, or the FDCA,
the PHSA, and regulations and guidance documents implementing these laws. The FDCA, PHSA and their corresponding regulations govern,
among other things, the testing, manufacturing, safety, purity, potency, labeling, packaging, storage, record keeping, distribution,
reporting, advertising and other promotional practices involving pharmaceutical products. Consent from the FDA is required before conducting
human clinical testing of drug products. FDA approval of a new drug application (NDA) or a biologics license application (BLA) also must
be obtained before marketing a new drug or biological product. The process of obtaining regulatory approvals and the subsequent compliance
with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial
resources.
U.S.
New Drug Product Development Process
Any
new drug or therapeutic biologic product must be approved by the FDA before it may be legally marketed in the United States. FDA approval
is also required before marketing an approved drug product for a new indication or condition of use. The process required by the FDA
before a new drug product candidate may be marketed in the United States generally involves the following:
| ● | Completion
of preclinical laboratory tests and in vivo studies in accordance with the FDA’s Good
Laboratory Practice (GLP) regulations and applicable requirements for the humane use of laboratory
animals or other applicable regulations; |
| ● | Submission
to the FDA of an investigational new drug (IND) application, which allows human clinical
trials to begin unless FDA objects (issues a “clinical hold”) within 30 calendar
days; |
| ● | Approval
by an independent institutional review board (IRB), reviewing each proposed clinical trial
and clinical site before each clinical trial may be initiated; |
| ● | Performance
of adequate and well-controlled human clinical trials in accordance with the protocol contained
in the approved IND and in accordance with the FDA’s Good Clinical Practice (GCP) regulations,
and any additional requirements for the protection of human research subjects and their health
information, to establish the safety and efficacy of the proposed product candidate for its
intended use; |
| ● | Preparation
and submission to the FDA of a new drug application (NDA) for marketing approval that includes
substantial evidence of safety and efficacy from results of nonclinical testing and clinical
trials; |
| ● | Review
of the product by an FDA advisory committee, if applicable; |
| ● | Satisfactory
completion of an FDA inspection of the manufacturing facility or facilities where the product
candidate is produced to assess compliance with current Good Manufacturing Practice (cGMP)
requirements and to assure that the facilities, methods and controls are adequate to preserve
the product candidate’s identity, safety, strength, quality, potency and purity; |
| ● | Potential
FDA audit of the nonclinical and clinical trial sites that generated the data in support
of the NDA; and |
| ● | Payment
of user fees and FDA review and approval of the NDA. |
The
testing and approval process of product candidates requires substantial time, effort, and financial resources. Satisfaction of the FDA’s
pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type,
complexity, and novelty of the product or disease. Before testing any product candidate in humans, the product candidate must undergo
preclinical testing. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry,
toxicity and formulation, as well as in vivo animal studies to assess the potential safety and activity of the product candidate and
to establish a rationale for therapeutic use. The conduct of the preclinical tests must comply with federal regulations and requirements
including GLPs.
Concurrent
with clinical trials, companies usually must complete some long-term preclinical testing, such as animal tests of reproductive adverse
events and carcinogenicity, and must also develop additional information about the chemistry and physical characteristics of the drug
and finalize a process for manufacturing the drug in commercial quantities in accordance with cGMP requirements. The manufacturing process
must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop
methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be
selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration
over its shelf life.
A
clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any
available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue
even after the IND is submitted. The IND automatically becomes active 30 calendar days after receipt by the FDA, unless before that time
the FDA raises concerns or questions related to a proposed clinical trial, including concerns that human research subjects will be exposed
to unreasonable health risks, and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve
any outstanding concerns before the clinical trial can begin. The FDA also may impose partial or full clinical holds on a product candidate
at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may
not begin, or recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that
submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, that issues arise that partially
or fully suspend or terminate such studies.
Human
Clinical Trials Under an IND
Clinical
trials involve the administration of the investigational product to healthy volunteers or patients under the supervision of qualified
investigators which generally are physicians not employed by, or under, the control of the trial sponsor. Clinical trials must be conducted
under written study protocols detailing, among other things, the objectives of the trial, subject selection and exclusion, the trial
procedures, the parameters to be used in monitoring safety, the criteria to be evaluated, and a statistical analysis plan. Each protocol
and any amendments to the protocol must be submitted to the FDA as part of the IND.
Further,
clinical trials must be conducted in accordance with federal regulations and GCP requirements, which include the requirements that all
research subjects provide their informed consent in writing for their participation in any clinical trial, as well as review and approval
by an IRB at each study site participating in the clinical trial or a central IRB. An IRB is charged with protecting the welfare and
rights of trial participants and considers items such as whether the risks to individuals participating in the clinical trials are minimized
and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must
be signed by each clinical trial subject, or their legal representative, reviews and approves the study protocol, and must monitor the
clinical trial until completed.
Human
clinical trials typically are conducted in three sequential phases that may overlap or be combined:
| ● | Phase
1. The product candidate initially is introduced into a small number of healthy human
subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion
and, if possible, to gain an early understanding of its value in treating patients. In the
case of some product candidates for severe or life-threatening diseases, especially when
the product candidate may be too inherently toxic to ethically administer to healthy volunteers,
the initial human testing is often conducted in patients. |
| ● | Phase
2. The product candidate is evaluated in a limited patient population to identify possible
adverse effects and safety risks, to preliminarily evaluate the efficacy of the product candidate
for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing
schedule. |
| ● | Phase
3. Phase 3 clinical trials are commonly referred to as “pivotal” or “registrational”
studies, which typically denotes a study which presents the data that the FDA or other relevant
regulatory agency will use to determine whether or not to approve a product. In Phase 3 studies,
the product candidate is administered to an expanded patient population, generally at multiple
geographically dispersed clinical trial sites in adequate and well-controlled clinical trials
to generate sufficient data to statistically demonstrate the efficacy and safety of the product
for approval. These clinical trials are intended to establish the overall risk/benefit ratio
of the product candidate and provide an adequate basis for product labeling. |
Post-approval
clinical trials, sometimes referred to as Phase 4 clinical trials, may be required by FDA, or may be voluntarily conducted after initial
approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication,
particularly for long-term safety follow-up.
During
all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical
data and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the
FDA.
Written
IND safety reports must be promptly submitted to the FDA and the investigators for: serious and unexpected adverse events; any findings
from other studies, in vivo laboratory tests or in vitro testing that suggest a significant risk for human subjects; or any clinically
important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The
sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting.
The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days
after the sponsor’s initial receipt of the information. Relevant additional information obtained by the sponsor that pertains to
a previously submitted IND safety report must be submitted as a follow-up IND safety report. Such report should be submitted within 15
calendar days after the sponsor receives the information.
Information
about certain clinical trials, including a description of the study and, in some cases, study results, must be submitted within specific
timeframes to the National Institutes of Health, or NIH, for public dissemination on their clinicaltrials.gov website. Manufacturers
or distributors of investigational products for the diagnosis, monitoring, or treatment of one or more serious or life-threatening diseases
or conditions where no other comparable or satisfactory therapeutic options exist must also have a publicly available policy on evaluating
and responding to requests for expanded access, sometimes called “compassionate use,” requests.
Additionally,
some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor that regularly
reviews accumulated data and advises the study sponsor regarding the continuing safety of the trial. This group, known as a Data and
Safety Monitoring Board (DSMB) or Data and Safety Monitoring Committee (DSMC), may also review interim data to assess the continuing
validity and scientific merit of the clinical trial. This group receives special access to unblinded data during the clinical trial and
may advise the sponsor to halt the clinical trial if it determined there is an unacceptable safety risk for subjects or on other grounds,
such as no demonstration of efficacy.
The
FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes
that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical
trial patients. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to
comply with the IRB’s requirements or if the trial poses an unexpected serious harm to subjects. The FDA or an IRB may also impose
conditions on the conduct of a clinical trial. Clinical trial sponsors may also choose to discontinue clinical trials as a result of
risks to subjects, a lack of favorable results, or changing business priorities.
Compliance
with current Good Manufacturing Practices (cGMP) Requirements
Manufacturers
of pharmaceutical and biological products must comply with applicable cGMP regulations, including quality control and quality assurance
and maintenance of records and documentation. Manufacturers and others involved in the manufacture and distribution of such products
also must register their establishments with the FDA and certain state agencies. Both domestic and foreign manufacturing establishments
must register and provide additional information to the FDA upon their initial participation in the manufacturing process. Establishments
may be subject to periodic, unannounced inspections by government authorities to ensure compliance with cGMP requirements and other laws.
Discovery of problems may result in a government entity placing restrictions on a product, manufacturer or holder of an approved NDA,
and may extend to requiring withdrawal of the product from the market. The FDA will not approve a NDA unless it determines that the manufacturing
processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within
required specification.
Concurrent
with clinical trials, companies usually complete additional preclinical studies and must also develop additional information about the
physical characteristics of the product candidate as well as finalize a process for manufacturing the product candidate in commercial
quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents or of causing other
adverse events with the use of small molecule products, the PHSA emphasizes the importance of manufacturing control for products whose
attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product
candidate and, among other requirements, the sponsor must develop methods for testing the identity, strength, quality, potency and purity
of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate
that the product candidate does not undergo unacceptable deterioration over its shelf life.
In
relation to the clinical trials that may be conducted in other countries with a view to obtaining a marketing authorization, there are
comparable cGMP requirements and other regulatory rules that are implemented nationally.
U.S.
FDA Review and Approval Process
Assuming
successful completion of the required clinical and preclinical testing, the results of the preclinical tests and clinical trials together
with detailed information relating to the product’s CMC, including negative or ambiguous results as well as positive findings,
and proposed labeling, among other things, are submitted to the FDA for NDA or BLA approval to market the product for one or more indications.
Under
the Prescription Drug User Fee Act (PDUFA), as amended, each NDA or BLA must be accompanied by a significant user fee. The FDA adjusts
the PDUFA user fees on an annual basis. The PDUFA also imposes an annual program fee for approved therapeutic products. Fee waivers or
reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small
business. Additionally, no user fees are assessed on NDAs for product candidates designated as orphan drugs, unless the product candidate
also includes a non-orphan indication.
In
addition, under the Pediatric Research Equity Act (PREA), an NDA for a new active ingredient, indication, dosage form, dosage regimen,
or route of administration, must contain data that are adequate to assess the safety and potential of the product for the claimed indications
in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product
is safe. Also, applications for product candidates intended for the treatment of adult cancer which are directed at molecular targets
that the FDA determines to be substantially relevant to the growth or progression of pediatric cancer, in place of the PREA investigations,
sponsors must submit, with the application, reports from molecularly targeted pediatric cancer investigations designed to yield clinically
meaningful pediatric study data, using appropriate formulations, to inform potential pediatric labeling. The FDA may, on its own initiative
or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product
for use in adults, or full or partial waivers from the pediatric data requirements. Orphan products are also exempt from the PREA requirements.
The
FDA initially reviews an NDA or BLA submission to determine if it is substantially complete before the agency accepts it for filing.
The FDA may refuse to file any NDA or BLA that it deems incomplete or not properly reviewable at the time of submission and may request
additional information. In that event, the NDA or BLA must be resubmitted with the additional information. The resubmitted application
also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth,
substantive review of the application.
The
FDA reviews the application to determine, among other things, whether the proposed product candidate is safe and effective ( or pure
and potent for BLAs) for its intended use, has an acceptable purity profile and whether the product candidate is being manufactured in
accordance with cGMP to assure and preserve the product candidate’s identity, safety, strength, quality, potency and purity. The
FDA may refer applications for novel therapeutic products or therapeutic products that present difficult questions of safety or efficacy
to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as
to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee,
but it considers such recommendations carefully when making decisions. During the product approval process, the FDA also will determine
whether a risk evaluation and mitigation strategy, (REMS) is necessary to assure the safe use of the product candidate. REMS use risk
minimization strategies beyond the professional labeling to ensure that the benefits of the product outweigh the potential risks. To
determine whether a REMS is needed, the FDA will consider the size of the population likely to use the product, seriousness of the disease,
expected benefit of the product, expected duration of treatment, seriousness of known or potential adverse events, and whether the product
is a new molecular entity. A REMS could include medication guides, physician communication plans and elements to assure safe use, such
as restricted distribution methods, patient registries and other risk minimization tools. If the FDA concludes a REMS is needed, the
sponsor of the NDA must submit a proposed REMS; the FDA will not approve the application without a REMS, if required.
Before
approving an application, the FDA will inspect the facilities at which the product candidate is manufactured. The FDA will not approve
the product candidate if it determines that the manufacturing processes and facilities are not in compliance with cGMP requirements or
otherwise are not adequate to assure consistent production of the product candidate within required specifications. Additionally, before
approving an application, the FDA typically will inspect one or more clinical sites to assure that the clinical trials were conducted
in compliance with IND trial requirements and GCP requirements.
On
the basis of the application and accompanying information, including the results of the inspection of the manufacturing facilities, the
FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the product with
specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission
and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies
have been addressed to the FDA’s satisfaction in a resubmission of the application, the FDA will issue an approval letter.
If
a product candidate receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications
for use may otherwise be limited. Further, the FDA may require that certain contraindications, warnings or precautions be included in
the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing or dispensing in the form of
a REMS, or otherwise limit the scope of any approval. The FDA may also require post-marketing clinical trials, sometimes referred to
as Phase 4 clinical trials, designed to further assess a product’s safety, and testing and surveillance programs to monitor the
safety of approved products that have been commercialized.
Every
five years, the FDA agrees to specified performance goals in the review of NDAs under the PDUFA. One such current goal is to review standard
NDAs in ten months after the FDA accepts the NDA for filing, and priority NDAs in six months, whereupon a review decision is to be made.
The FDA does not always meet its PDUFA goal dates for standard and priority NDAs and its review goals are subject to change from time
to time. The review process and rhe PDUFA goal date may be extended by three months if the FDA requests or the NDA sponsor otherwise
provides additional information or clarification regarding information already provided in the submission within the last three months
before the PDUFA goal date.
Post-Approval
Requirements
After
approval, there also are continuing annual program user fee requirements for approved products, excluding, under certain circumstances,
orphan products.
Rigorous
and extensive FDA regulation of pharmaceutical and biological products continues after approval, particularly with respect to cGMP requirements.
Manufacturers are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance
and maintenance of records and documentation. To help reduce the increased risk of the introduction of adventitious agents, the PHSA
emphasizes the importance of manufacturing controls for products whose attributes cannot be precisely defined. The PHSA also provides
authority to the FDA to immediately suspend licenses in situations where there exists a danger to public health, to prepare or procure
products in the event of shortages and critical public health needs, and to authorize the creation and enforcement of regulations to
prevent the introduction or spread of communicable diseases in the United States and between states.
Other
post-approval requirements applicable to pharmaceutical products include reporting of cGMP deviations that may affect the identity, potency,
purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety
and efficacy information and complying with electronic record and signature requirements. In addition, the FDA conducts laboratory research
related to the regulatory standards on the safety, purity, and potency of pharmacological products.
In
addition, manufacturers and other entities involved in the manufacture and distribution of approved pharmaceuticals are required to register
their establishments with the FDA and certain state agencies, list their products, and are subject to periodic announced and unannounced
inspections by the FDA and these state agencies for compliance with current cGMP and other requirements, which impose certain procedural
and documentation requirements upon us and third-party manufacturers. Manufacturers must continue to expend time, money, and effort in
the areas of production and quality-control to maintain compliance with current cGMPs. Regulatory authorities may withdraw product approvals
or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing,
or if previously unrecognized problems are subsequently discovered. In addition, changes to the manufacturing process or facility generally
require prior FDA approval or notification before being implemented, and other types of changes to the approved product, such as adding
new indications and additional labeling claims, are also subject to further FDA review and approval.
Moreover,
the Drug Quality and Security Act imposes obligations on manufacturers of pharmaceutical products related to product tracking and tracing.
Adverse
event reporting and submission of periodic reports, including annual reports and deviation reports, are required following FDA approval
of a NDA or BLA. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or
frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in significant regulatory actions.
Such actions may include refusal to approve pending applications, license suspension or revocation, imposition of a partial or full clinical
hold or termination of clinical trials, warning letters, untitled letters, modification of promotional materials or labeling, provision
of corrective information, imposition of post-market requirements including the need for additional testing, imposition of distribution
or other restrictions under a REMS, product recalls, product seizures or detentions, refusal to allow imports or exports, total or partial
suspension of production or distribution, FDA debarment, injunctions, fines, consent decrees, corporate integrity agreements, suspension
and debarment from government contracts, and refusal of orders under existing government contracts, exclusion from participation in federal
and state healthcare programs, restitution, disgorgement, or civil or criminal penalties, including fines and imprisonment, and result
in adverse publicity, among other adverse consequences.
A
sponsor also must comply with the FDA’s advertising and promotion requirements, such as the prohibition on promoting products for
uses or inpatient populations that are inconsistent with the product’s approved labeling (known as “off-label use”).
The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that
is found to have improperly promoted off-label uses may be subject to significant liability. Violations relating to the promotion of
off-label uses may lead to investigations alleging violations of federal and state healthcare fraud and abuse and other laws, as well
as state consumer protection laws. Companies, however, may generally share truthful and non-misleading information that is otherwise
consistent with a product’s FDA approved labeling. Discovery of previously unknown problems or the failure to comply with the applicable
regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well
as possible civil or criminal sanctions.
Failure
to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval,
may subject an applicant or manufacturer to administrative or judicial civil or criminal actions and adverse publicity. These actions
could include refusal to approve pending applications or supplemental applications, withdrawal of an approval, clinical hold, suspension
or termination of a clinical trial by an IRB, warning or untitled letters, product recalls, product seizures, total or partial suspension
of production or distribution, injunctions, fines or other monetary penalties, refusals of government contracts, mandated corrective
advertising or communications with healthcare providers, debarment, restitution, disgorgement of profits or other civil or criminal penalties.
Broadly
equivalent requirements and controls typically apply in other countries to the submission of marketing authorization applications and,
post-approval, to the holding of such marketing authorizations.
Other
Healthcare Laws and Regulations
Our
business activities, including but not limited to, research, sales, promotion, distribution, medical education, and other activities
following product approval will be subject to regulation by numerous federal and state regulatory and law enforcement authorities in
the United States in addition to the FDA, including potentially the Department of Justice, the Department of Health and Human Services
(HHS) and its various divisions, including the Office of Inspector General, the Centers for Medicare & Medicaid Services (CMS) and
the Health Resources and Services Administration, the Department of Veterans Affairs, the Department of Defense, and state and local
governments. Healthcare providers and third-party payors play a primary role in the recommendation and use of pharmaceutical products
that are granted marketing approval. Arrangements with third-party payors, existing or potential customers and referral sources, including
healthcare providers, are subject to broadly applicable fraud and abuse laws and regulations, and these laws and regulations may constrain
the business or financial arrangements and relationships through which manufacturers conduct clinical research, market, sell and distribute
the products for which they obtain marketing approval. Such restrictions under applicable federal and state healthcare laws and regulations
include the following:
| ● | The
federal Anti-Kickback Statute, which prohibits, among other things, persons and entities
from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly
or indirectly, in cash or kind, in exchange for, or to induce, either the referral of an
individual for, or the purchase, order or recommendation of, any good or service for which
payment may be made under federal healthcare programs such as the Medicare and Medicaid programs.
This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers,
on the one hand, and prescribers, purchasers, formulary managers and other individuals and
entities on the other. The Patient Protection and Affordable Care Act, as amended by the
Health Care and Education Reconciliation Act (collectively, the ACA) amended the intent requirement
of the federal Anti-Kickback Statute such that a person or entity no longer needs to have
actual knowledge of this statute or specific intent to violate it in order to commit a violation; |
| ● | The
federal civil and criminal false claims, including the civil FCA, and Civil Monetary Penalties
Laws which prohibit, among other things, individuals or entities from knowingly presenting,
or causing to be presented, claims for payment from Medicare, Medicaid or other third-party
payors that are false or fraudulent, or making a false statement to avoid, decrease, or conceal
an obligation to pay money to the federal government. Certain marketing practices, including
off-label promotion, also may implicate the FCA. In addition, the ACA codified case law that
a claim including items or services resulting from a violation of the federal Anti-Kickback
Statute constitutes a false or fraudulent claim for purposes of the FCA; |
| ● | The
federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices,
therapeutic products and medical supplies for which payment is available under Medicare,
Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to
report annually to the Centers for Medicare & Medicaid Services, or the CMS, information
related to payments and other transfers of value made to physicians (defined to include doctors,
dentists, optometrists, podiatrists and chiropractors), certain other healthcare professionals
(such as physician assistants and nurse practitioners), and teaching hospitals, and ownership
and investment interests held by physicians and other healthcare providers and their immediate
family members; |
| ● | Health
Insurance Portability and Accountability Act of 1996 (HIPAA) prohibits knowingly and willfully
executing, or attempting to execute, a scheme to defraud or to obtain, by means of false
or fraudulent pretenses, representations or promises, any of the money or property owned
by, or under the custody or control of, a healthcare benefit program, regardless of whether
the payor is public or private, in connection with the delivery or payment for health care
benefits, knowingly and willfully embezzling or stealing from a health care benefit program,
willfully obstructing a criminal investigation of a health care offense and knowingly and
willfully falsifying, concealing, or covering up by any trick or device a material fact or
making any materially false statements in connection with the delivery of, or payment for,
healthcare benefits, items, or services relating to healthcare matters. Additionally, the
ACA amended the intent requirement of certain of these criminal statutes under HIPAA so that
a person or entity no longer needs to have actual knowledge of the statute, or the specific
intent to violate it, to have committed a violation; and |
| ● | State
and foreign law equivalents of each of the above federal laws, such as anti-kickback and
false claims laws which may apply to items or services reimbursed by any third-party payor,
including commercial insurers; state laws that require pharmaceutical companies to comply
with the pharmaceutical industry’s voluntary compliance guidelines and the relevant
compliance guidance promulgated by the federal government or otherwise restrict payments
that may be made to healthcare providers and other potential referral sources; state laws
that require drug manufacturers to report information related to payments and other transfers
of value to physicians and other healthcare providers and drug pricing and/or marketing expenditures;
and state and local laws requiring the registration of pharmaceutical sales representatives
and state laws governing the privacy and security of health information in certain circumstances,
many of which differ from each other in significant ways and may not have the same effect,
thus complicating compliance efforts. |
Further,
we may be subject to data privacy and security regulation by both the federal government and the states and foreign jurisdictions in
which we conduct our business. HIPAA, as amended by the Health Information Technology for Clinical Health Act of 2009 (HITECH), and its
respective implementing regulations imposes certain requirements, including mandatory contractual terms, on covered entities, business
associates and their covered subcontractors relating to the privacy, security, and transmission of certain individually identifiable
health information known as protected health information. Among other things, HITECH, through its implementing regulations, makes HIPAA’s
security standards and certain privacy standards directly applicable to business associates, defined as a person or organization, other
than a member of a covered entity’s workforce, that creates, receives, maintains, or transmits protected health information on
behalf of a covered entity for a function or activity regulated by HIPAA. HITECH also strengthened the civil and criminal penalties that
may be imposed against covered entities, business associates, subcontractors, and individuals, and gave state attorneys general new authority
to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and
costs associated with pursuing federal civil actions. In addition, other federal and state laws may govern the privacy and security of
health and other information in certain circumstances, many of which differ from each other in significant ways and may not be pre-empted
by HIPAA, thus complicating compliance efforts.
To
the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may
include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation
of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
In
the EU, the data privacy laws are generally perceived to be stricter than those which apply in the United States and include specific
requirements for the transfer of personal data outside the EU to the United States to ensure that EU standards of data privacy will be
applied to such data.
Violation
of the laws described above or any other governmental laws and regulations may result in significant penalties, including administrative,
civil and criminal penalties, damages, fines, the curtailment or restructuring of operations, the exclusion from participation in federal
and state healthcare programs, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, imprisonment,
and additional reporting requirements and oversight if a person becomes subject to a corporate integrity agreement or similar agreement
to resolve allegations of non-compliance with these laws. Furthermore, efforts to ensure that business activities and business arrangements
comply with applicable healthcare laws and regulations can be costly for manufacturers of branded prescription products.
Coverage
and Reimbursement
Significant
uncertainty exists as to the coverage and reimbursement status of any products for which we may obtain regulatory approval. In the United
States, sales of any product candidates for which regulatory approval for commercial sale is obtained will depend in part on the availability
of coverage and adequate reimbursement from third-party payors. Third-party payors include government authorities and health programs
in the United States such as Medicare and Medicaid, managed care providers, private health insurers and other organizations. These third-party
payors are increasingly reducing reimbursements for medical products and services. The process for determining whether a payor will provide
coverage for a drug product may be separate from the process for setting the reimbursement rate that the payor will pay for the drug
product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all
of FDA-approved drugs for a particular indication. Additionally, the containment of healthcare costs has become a priority of federal
and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign
governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement
and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more
restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results.
A
payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further,
coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process
is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products
to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the
first instance.
Third-party
payors are increasingly challenging the price and examining the medical necessity and cost of medical products and services, in addition
to their safety and efficacy. New metrics frequently are used as the basis for reimbursement rates, such as average sales price, average
manufacturer price and actual acquisition cost. In order to obtain coverage and reimbursement for any product that might be approved
for sale, it may be necessary to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and rational
of the cost of the products, in addition to the costs required to obtain regulatory approvals. If third-party payors do not consider
a product to be cost saving when compared to other available therapies, they may not cover the product after approval as a benefit under
their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit.
The
marketability of any product candidates for which we or our collaborators receive regulatory approval for commercial sale may suffer
if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in
the United States has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and
third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more
products for which we or our collaborators receive regulatory approval, less favorable coverage policies and reimbursement rates may
be implemented in the future. The cost containment measures that healthcare payors and providers are instituting and any healthcare reform
could significantly reduce our revenues from the sale of any approved product candidates. We cannot provide any assurances that we will
be able to obtain and maintain third-party coverage or adequate reimbursement for our product candidates in whole or in part.
In
the EU, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only
after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost of
a particular product candidate to currently available therapies. EU member states may approve a specific price for a product or it may
instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other member
states allow companies to fix their own prices for products, but monitor and control company profits. The downward pressure on health
care costs has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in
some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any
country that has price controls or reimbursement limitations may not allow favorable reimbursement and pricing arrangements.
European
and Other Regulatory Approval
Whether
or not FDA approval has been obtained, approval of a product by comparable regulatory authorities in Europe, Japan and other countries
will likely be necessary prior to commencement of marketing such product in such countries. The regulatory authorities in each country
may impose their own requirements and may refuse to grant an approval, or may require additional data before approving it, even though
the relevant product has been approved by the FDA or another authority. The regulatory authorities in the European Union (“EU”),
Australia and other developed countries have lengthy approval processes for pharmaceutical products. The process for gaining an approval
in a particular country may vary from the process in another country, but generally follows a similar sequence to that described for
FDA approval. The EU has established a European agency for the evaluation of medical products, with both a centralized community procedure
and a decentralized procedure, the latter being based on the principle of licensing within one member country followed by mutual recognition
by the other member countries.
The
process of obtaining approval for a new drug in Japan resembles U.S. and EU procedures in both substance and scope. All NDAs are collected
and reviewed by the Japanese Pharmaceuticals and Medical Devices Agency, or PMDA. PMDA review typically involves at least two formal
evaluations to establish the safety and efficacy of the drug candidate, as well as one cGMP facility inspection. Consultations to correct
outstanding issues are conducted as needed. Assuming satisfactory results, these reports are communicated to the Ministry of Health,
Labour, and Welfare, or MHLW, which then issues a final approval of the drug.
Approval
of Biosimilars
The
pathway for approval of biosimilar products was established by the Biologics Price Competition and Innovation Act of 2009, or BPCIA,
enacted on March 23, 2010, as part of the Patient Protection and Affordable Care Act. The BPCIA established this abbreviated pathway
under section 351(k) of the Public Health Service Act, or PHSA. Subsequent to the enactment of the BPCIA, the FDA issued draft guidance
regarding the demonstration of biosimilarity as well as the submission and review of biosimilar applications. Biosimilars are to be compared
for biosimilarity to “Reference Products” already licensed by the FDA. Market success of biosimilar products will depend
on demonstrating to patients, physicians, payors and relevant authorities that such products are similar in quality, safety and efficacy
as compared to the reference product. The BPCIA requires a biosimilar applicant to demonstrate biochemical and biologic biosimilarity
using clinical studies with respect to the reference product that has been approved by FDA in the United States. Biosimilars approved
in the EU and other non-U.S. jurisdictions may not be approved in the United States without additional studies demonstrating biosimilarity
to an FDA-approved reference product. Biosimilars approved in the United States may also not be approved in foreign jurisdictions without
additional bridging studies. We will continue to analyze and incorporate into our biosimilar development plans all final regulations
and guidance issued by the FDA, pharmacy substitution policies enacted by state governments and other applicable requirements established
by relevant authorities.
A
biosimilars approval pathway has been in place in the E.U. since 2003. The EMA has issued a number of scientific and product specific
biosimilar guidelines, including requirements for approving biosimilars containing monoclonal antibodies. In the E.U., biosimilars are
generally approved under the centralized procedure. The approval pathway allows sponsors of a biosimilar to seek and obtain regulatory
approval based in part on reliance on the clinical trial data of an innovator product to which the biosimilar has been demonstrated,
through comprehensive comparability studies, to be “similar.” In many cases, this allows biosimilars to be brought to market
without conducting the full complement of clinical trials typically required for novel biologic drugs.
Employees
As
of August 30, 2024, ANEW has three (3) full-time employees and utilizes the services of four consultants.
Properties
The
Company is currently using three (3) remote offices in Charlotte, NC, Omaha, NE and Scottsdale, AZ, each consisting of approximately
400 sq ft square feet, and provided for the Company’s use rent-free by the Company’s employees.
DIVIDEND
POLICY
We
have not declared or paid dividends on our common shares since our formation, and we do not anticipate paying dividends in the foreseeable
future.
Instead,
we will retain any earnings for use in our business. This policy will be reviewed by our board of directors from time to time in
light of, among other things, our earnings and financial position.
No
distribution may be made if, after giving it effect, we would not be able to pay its debts as they become due in the usual course of
business; or the corporation’s total assets would be less than the sum of its total liabilities plus (unless the articles of incorporation
permit otherwise) the amount that would be needed, if we were to be dissolved at the time of the distribution, to satisfy the preferential
rights upon dissolution of shareholders whose preferential rights are superior to those receiving the distribution.
The
board of directors may base a determination that a distribution is not prohibitive either on consolidated financial statements prepared
on the basis of accounting practices and principles that are reasonable in the circumstances or on a fair valuation of other method that
is reasonable in the circumstances.
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF
FINANCIAL
CONDITION AND RESULTS OF OPERATIONS
Except
where the context otherwise requires or where otherwise indicated, references in this section to the terms “ANEW,” “we,”
“us,” “our,” “our Company” and “our business” refer, to ANEW Medical, Inc., a Delaware
corporation together with its consolidated subsidiaries.
Forward
Looking Statements
All
statements other than statements of historical facts contained in this report, including statements regarding future operations, are
forward-looking statements. In some cases, forward-looking statements may be identified by words such as “believe,” “may,”
“will,” “estimate,” “continue,” “anticipate,” “intend,” “could,”
“would,” “expect,” “objective,” “plan,” “potential,” “seek,”
“grow,” “target,” “if,” and similar expressions intended to identify forward-looking statements.
We have based these forward-looking statements largely on our current expectations and projections about future events and trends that
we believe may affect our financial condition, results of operations, business strategy, short-term and long-term business operations,
objectives, and financial needs.
Overview
ANEW
seeks to develop essential medicines for the treatment of chronic diseases — cancer, cardiovascular, muscle, skin, and neurodegenerative
disorders. ANEW currently has two platform technologies. One platform consists of proprietary, patented technology programs that include
(a) a gene therapy program that uses a gene therapy approach to produce a therapeutic protein called “Klotho” inside the
body to treat neurodegenerative diseases and other diseases of aging, and (b) a library of melanocortin receptor-binding molecules that
activate or inactive melanocortin receptors in the body. ANEW’s second platform is an “off-patent” portfolio of generic
drugs and biosimilar biologics selling hard-to source, difficult to find generic drugs and off-patent biologic therapies.
The
Business Combination
On
May 30, 2023, the Company, then known as Redwoods Acquisition Corp. and a newly formed wholly-owned Wyoming subsidiary of Redwoods
Acquisition Corp. (“Merger Sub”) entered into a Business Combination Agreement with ANEW Medical, Inc., a Wyoming corporation
(“ANEW Wyoming”). On June 21, 2024, at the closing of the Business Combination Agreement, Merger Sub merged with and into
ANEW Wyoming and ANEW Wyoming become a wholly-owned subsidiary of Redwoods. At the same time, the name of the Company was changed to
ANEW Medical, Inc.
The
business combination was accounted for as a reverse recapitalization. ANEW Wyoming was deemed the accounting predecessor and the combined
entity is the successor SEC registrant, meaning that the ANEW Wyoming’s financial statements for previous periods will be disclosed
in the registrant’s future periodic reports filed with the SEC.
Components
of Our Results of Operations
We
have been a research and development stage company and our historical results may not be indicative of our future results for reasons
that may be difficult to anticipate. Accordingly, the drivers of our future financial results, as well as the components of such results,
may not be comparable to our historical or projected results of operations.
Management’s
Discussion and Analysis of Financial Condition and Results of Operations
References
in this Registration Statement to “we,” “us” or the “Company” refer to ANEW Medical, Inc. References
to our “management” or our “management team” refer to our officers and directors. The following discussion and
analysis of the Company’s financial condition and results of operations should be read in conjunction with the unaudited condensed
consolidated financial statements and the notes thereto contained elsewhere in this Quarterly Report. Certain information contained in
the discussion and analysis set forth below includes forward-looking statements that involve risks and uncertainties.
Special
Note Regarding Forward-Looking Statements
This
Registration Statement includes “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933,
as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange
Act”), that are not historical facts and involve risks and uncertainties that could cause actual results to differ materially from
those expected and projected. All statements, other than statements of historical fact included in this Quarterly Report, including,
without limitation, statements in this “Management’s Discussion and Analysis of Financial Condition and Results of Operations”
regarding the search for an initial business combination, the Company’s financial position, business strategy and the plans and
objectives of management for future operations, are forward-looking statements. Words such as “expect,” “believe,”
“anticipate,” “intend,” “estimate,” “seek” and variations and similar words and expressions
are intended to identify such forward-looking statements. Such forward-looking statements relate to future events or future performance,
but reflect management’s current beliefs, based on information currently available. A number of factors could cause actual events,
performance or results to differ materially from the events, performance and results discussed in the forward-looking statements. For
information identifying important factors that could cause actual results to differ materially from those anticipated in the forward-looking
statements, please refer to the Risk Factors section of the Company’s final prospectus for its initial public offering filed with
the U.S. Securities and Exchange Commission (the “SEC”). The Company’s filings with the SEC can be accessed on the
EDGAR section of the SEC’s website at www.sec.gov. Except as expressly required by applicable securities law, the Company disclaims
any intention or obligation to update or revise any forward-looking statements whether as a result of new information, future events
or otherwise.
Overview
ANEW
Medical, Inc. (the “Company” or “ANEW”) develops essential medicines for the treatment of chronic diseases –
cancer, cardiovascular, and neurodegenerative disorders. The Company currently has acquired two licensed platforms a generic drug portfolio
and a biosimilar biologics platform that uses biologic therapies to treat cancer, and two proprietary, patented technologies involving
the melanocortin receptor-binding molecules and a gene therapy platform which uses a gene therapy approach to introduce a therapeutic
protein called “Klotho” inside the body to treat neurodegenerative diseases.
On
May 30, 2023, the Company, then known as Redwoods Acquisition Corp. entered into a business combination agreement (the “Business
Combination Agreement”) with ANEW Medical, Inc., a Wyoming corporation. Then, on June 21, 2024, the closing of the Business Combination
Agreement occurred resulting in ANEW Medical, Inc., a Wyoming corporation becoming a wholly- owned subsidiary of the Company, and the
Company simultaneously changed its name to ANEW Medical, Inc.
Critical
Accounting Policies and Estimates
The
preparation of unaudited condensed consolidated financial statements and related disclosures in conformity with accounting principles
generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts
of assets and liabilities, disclosure of contingent assets and liabilities at the date of the unaudited condensed consolidated financial
statements, and income and expenses during the periods reported. Actual results could materially differ from those estimates. We have
not identified any critical accounting estimates; we have identified the following critical accounting policies:
Fair
Value of Financial Instruments
FASB
ASC Topic 820 “Fair Value Measurements and Disclosures” defines fair value, the methods used to measure fair value and the
expanded disclosures about fair value measurements. Fair value is the price that would be received to sell an asset or paid to transfer
a liability in an orderly transaction between the buyer and the seller at the measurement date. In determining fair value, the valuation
techniques consistent with the market approach, income approach and cost approach shall be used to measure fair value. FASB ASC Topic
820 establishes a fair value hierarchy for inputs, which represent the assumptions used by the buyer and seller in pricing the asset
or liability. These inputs are further defined as observable and unobservable inputs. Observable inputs are those that buyer and seller
would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs
reflect the Company’s assumptions about the inputs that the buyer and seller would use in pricing the asset or liability developed
based on the best information available in the circumstances.
The
fair value hierarchy is categorized into three levels based on the inputs as follows:
| Level
1 - |
Valuations
based on unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access.
Valuation adjustments and block discounts are not being applied. Since valuations are based on quoted prices that are readily and
regularly available in an active market, valuation of these securities does not entail a significant degree of judgment. |
| |
|
| Level
2 - |
Valuations
based on (i) quoted prices in active markets for similar assets and liabilities, (ii) quoted prices in markets that are not active
for identical or similar assets, (iii) inputs other than quoted prices for the assets or liabilities, or (iv) inputs that are derived
principally from or corroborated by market through correlation or other means. |
| |
|
| Level
3 - |
Valuations
based on inputs that are unobservable and significant to the overall fair value measurement. |
The
fair value of the Company’s certain assets and liabilities, which qualify as financial instruments under ASC Topic 820, “Fair
Value Measurements and Disclosures,” approximates the carrying amounts represented in the consolidated balance sheet. The fair
values of cash and cash equivalents, and other current assets, accrued expenses, due to sponsor are estimated to approximate the carrying
values as of March 31, 2024 and December 31, 2023 due to the short maturities of such instruments. See Note 9 to unaudited condensed
consolidated financial statements for the disclosure of the Company’s assets and liabilities that were measured at fair value on
a recurring basis.
The
fair value of the Company’s certain assets and liabilities, which qualify as financial instruments under ASC Topic 820, “Fair
Value Measurements and Disclosures,” approximates the carrying amounts represented in the consolidated balance sheet. The fair
values of cash and cash equivalents, and other current assets, accrued expenses, due to sponsor are estimated to approximate the carrying
values as of March 31, 2024 and December 31, 2023 due to the short maturities of such instruments. See Note 9 for the disclosure of the
Company’s assets and liabilities that were measured at fair value on a recurring basis.
Convertible
Promissory Notes
The
Company initially accounted for its convertible promissory notes under ASC 815, “Derivatives and Hedging” and elected the
fair value option under ASC 825. Using the fair value option method, each convertible promissory note is required to be recorded at its
initial fair value on the date of issuance, and each balance sheet date thereafter. Changes in the estimated fair value of the notes
are recognized as a non-cash gain or loss on the statements of operations.
Subsequently,
the conversion feature of the convertible promissory notes was amended on May 15, 2023; the holder of the convertible promissory notes,
in its sole discretion, may convert any or all of the unpaid principal under the convertible promissory notes into common stocks of the
Company (see Note 6). As a result, the Company assessed the change in conversion feature and determined that the convertible promissory
notes should be recorded as debt (liability) at cash proceeds on the balance sheet. The Company’s assessment of the embedded conversion
feature considered the derivative scope exception guidance under ASC 815 pertaining to equity classification of contracts in an entity’s
own equity.
The
Company’s assessment was also based on ASC 470-50 - Debt Modifications and Exchanges; management determined that the amended conversion
option (which is based on shares of the Company’s common stocks) is substantially different from the original conversion option
(which was based on units). Since each unit consists of one share of common stock, one share of right convertible into one-tenth (1/10)
of one share of common stock upon the consummation of a Business Combination, the original conversion option offers at least 10% more
shares of common stock (including underlying shares from the rights conversion) than the amended conversion option. As such, a remeasurement
under ASC 825 has occurred and the previously selected fair value option is no longer applied. The convertible promissory notes were
recorded as debt (liability) at cash proceeds on the balance sheet effective May 15, 2023.
For
all newly issued and unmodified convertible promissory notes, the Company elects an early adoption of the Financial Accounting Standards
Board (“FASB”) issued Accounting Standards Update (“ASU”) 2020-06, Debt - Debt with Conversion and Other Options
(Subtopic 470-20) and Derivatives and Hedging - Contracts in Entity’s Own Equity (Subtopic 815-40) (“ASU 2020-06”)
and accounts for newly issued as debt (liability) on the balance sheet. The Company considers the derivative scope exception guidance
under ASC 815 pertaining to equity classification of contracts in an entity’s own equity.
Warrants
The
Company accounts for warrants (Public Warrants or Private Warrants) as either equity-classified or liability-classified instruments based
on an assessment of the warrant’s specific terms and applicable authoritative guidance in Financial Accounting Standards Board
(“FASB”) Accounting Standards Codification (“ASC”) 480, Distinguishing Liabilities from Equity (“ASC 480”)
and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial
instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements
for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock and whether
the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control,
among other conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the
time of warrant issuance and as of each subsequent quarterly period end date while the warrants are outstanding.
For
issued or modified warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component
of equity at the time of issuance. For issued or modified warrants that do not meet all the criteria for equity classification, the warrants
are required to be recorded as liabilities at their initial fair value on the date of issuance, and each balance sheet date thereafter.
Changes in the estimated fair value of the warrants are recognized as a non-cash gain or loss on the statements of operations. The Company
has elected to account for its Public Warrants as equity and the Private Warrants as liabilities.
Net
Income (Loss) Per Share
The
Company complies with accounting and disclosure requirements of FASB ASC 260, Earnings Per Share. In order to determine the net income
(loss) attributable to both the redeemable shares and non-redeemable shares, the Company first considered the undistributed income (loss)
allocable to both the redeemable shares and non-redeemable shares and the undistributed income (loss) is calculated using the total net
loss less any dividends paid. We then allocated the undistributed income (loss) ratably based on the weighted average number of shares
outstanding between the redeemable and non-redeemable shares. Any re-measurement of the accretion to redemption value of the common stock
subject to possible redemption was considered to be dividends paid to the public stockholders.
Results
of Operations
For
accounting purposes, the Business Combination are treated as a reverse acquisition and, as such, the historical financial statements
of the accounting acquirer, ANEW Medical, Inc., a Wyoming corporation, became the historical financial statements of publicly traded
ANEW Medical, Inc., a Delaware corporation. The Results of Operations herein are those of the accounting acquirer, ANEW Medical, Inc.,
a Wyoming corporation.
We
have not generated any operating revenues to date. To date, the Company’s operations have consisted of acquiring our licensed platforms
and patents, and planning for the Business Combination. We incur expenses as a result of being a public company (for legal, financial
reporting, accounting and auditing compliance), as well as our expenses associated with planning our research and clinical testing operations.
Results
of Operations for the year ended December 31, 2023 Compared to the year ended December 31, 2022.
Revenues
We
have not earned any revenue since our inception.
Operating
Expenses
Operating
expenses are composed of consultant fees and professional fees.
Our
operating expenses for the year ended December 31, 2023 were $631,322 compared to $574,374 for
the year ended December 31, 2022, an increase of $56,948 or 9.9%. The increase was primarily
due to increased expenses associated with preparing for our business combination including increases in third party consulting fees and
professional fees.
Net
Loss
For
the year ended December 31, 2023, we incurred a net loss of $631,322 compared to a net loss
of $574,374 for the year ended December 31, 2022, an increase of $56,948 or 9.9%. The increase
in net loss was primarily due to increased expenses associated with preparing for our business combination including increases in third
party consulting fees and professional fees.
Liquidity
and Capital Resources
| | |
For the Year
Ended | | |
| | |
| |
| | |
December, | | |
Change | |
| | |
2023 | | |
2022 | | |
$ | | |
% | |
| | |
| | |
| | |
| | |
| |
| Net cash (used in) operating activities | |
$ | (707,458 | ) | |
$ | (598,593 | ) | |
$ | (108,865 | ) | |
| 18.2 | % |
| Net cash provided by
financing activities | |
| 374,000 | | |
| 500,000 | | |
| (126,000 | ) | |
| 25.2 | % |
| Net increase (decrease) in cash and cash equivalents | |
| (73,064 | ) | |
| (224,981 | ) | |
| 151,917 | | |
| 67.5 | % |
| Cash, beginning of year | |
| 75,872 | | |
| 300,853 | | |
| (224,981 | ) | |
| 74.8 | % |
| Cash, end of period | |
$ | 2,808 | | |
$ | 75,872 | | |
$ | (73,064 | ) | |
| 96.2 | % |
Operating
activities
Net
cash used in operating activities for the year ended December 31, 2023 was $707,458 compared to $598,593 for the year ended December
31, 2022, an increase of $108,865 or 18.2%. The significant increase in cash used in operating activities is primarily attributable
to increases in expenses as we prepared to close our business combination. We expect net cash from operating activities to be negative
in the coming periods, until the Company’s products are able to produce meaningful revenue.
Financing
activities
Net
cash provided by financing activities for the year ended December 31, 2023 was $374,000, all
from the sale of convertible promissory notes and the Company’s common stock and promissory notes to investors.
Results
of Operations for the Three and Six Months Ended June 30, 2024 Compared to the Three and Six Months Ended June 30, 2023
Revenues
We
have not earned any revenue since our inception.
Operating
Expenses
Operating
expenses are composed of consultant fees and professional fees.
Our
operating expenses for the three months ended June 30, 2024 were $395,607 compared to $244,252 for the three months ended June 30, 2023,
an increase of $151,355 or approximately 62%. The increase was primarily due to increased expenses associated with preparing for our
business combination including increases in third party consulting fees and professional fees.
Our
operating expenses for the six months ended June 30, 2024 were $817,652 compared to $396,852 for the six months ended June 30, 2023,
an increase of $420,800 or approximately 106%. The increase was primarily due to increased expenses associated with preparing for our
business combination including increases in third party consulting fees and professional fees.
Net
Loss
For
the three months ended June 30, 2024, we incurred a net loss of $451,639 compared to a net loss of $264,389 for the three month period
ended June 30, 2023, an increase of $187,250 or approximately 71%. The increase in net loss was primarily due to increased expenses associated
with preparing for our business combination including increases in third party consulting fees and professional fees.
For
the six months ended June 30, 2024, we incurred a net loss of $1,123,683 compared to a net loss of $436,872 for the six month period
ended June 30, 2023, an increase of $686,811 or approximately 157%. The increase in net loss was primarily due to increased expenses
associated with preparing for our business combination including increases in third party consulting fees and professional fees.
Liquidity
and Capital Resources
| | |
For the Six
Months Ended | | |
| | |
| |
| | |
June
30, | | |
Change | |
| | |
2024 | | |
2023 | | |
$ | | |
% | |
| | |
| | |
| | |
| | |
| |
| Net cash used in operating activities | |
$ | (929,399 | ) | |
$ | (265,429 | ) | |
$ | (663,970 | ) | |
| 250 | % |
| Net cash used in investing activities | |
| (123,497 | ) | |
| (20,000 | ) | |
| (103,497 | ) | |
| 517 | % |
| Net cash provided by
financing activities | |
| 1,895,424 | | |
| 250,000 | | |
| 1,645,424 | | |
| 658 | % |
| Net increase (decrease) in cash and cash equivalents | |
| 842,528 | | |
| (35,429 | ) | |
| 877,957 | | |
| (2,478 | )% |
| Cash, beginning of year | |
| 2,808 | | |
| 75,872 | | |
| (73,064 | ) | |
| (96 | )% |
| Cash, end of period | |
$ | 845,336 | | |
$ | 40,443 | | |
$ | 804,893 | | |
| 1,990 | % |
Operating
Activities
Net
cash used in operating activities for the six months ended June 30, 2024 was $929,399, compared to $265,429, for the six months ended
June 30, 2023, an increase of $663,970 or approximately 250%. The significant increase in cash used in operating activities is primarily
attributable to increases in expenses as we prepared to close our business combination. We expect net cash from operating activities
to be negative in the coming periods, until our products are able to produce meaningful revenue.
Investing
Activities
Net
cash used in investing activities for the six months ended June 30, 2024 was $123,497, compared to $20,000 for the six months ended June
30, 2023, an increase of $103,497 or approximately 517%. The increase in cash used in investing activities is primarily attributable
to licensing payments made in the period.
Financing
Activities
Net
cash provided by financing activities for the six months ended June 30, 2024 was $1,895,424, which consisted of investments and proceeds
from the Business Combination. For the six months ended June 30, 2023, net cash provided by financing activities was $250,000, primarily
from the sale of the Company’s common stock and promissory notes to investors.
Liquidity,
Capital Resources and Going Concern
As
of June 30, 2024, the Company had cash of $845,336 and net working capital of $101,273.
The
Company has incurred and expects to continue to incur significant professional costs to remain as a publicly traded company and incurred
significant transaction costs related to the consummation of the Business Combination.
The
accompanying consolidated financial statements have been prepared as if the Company will continue as a going concern. The Company has
incurred significant operating losses and negative cash flows from operations since inception. As of June 30, 2024, the Company had cash
of approximately $845,000 and an accumulated deficit of approximately $5.5 million. The Company has incurred recurring losses,
has experienced recurring negative operating cash flows, and requires significant cash resources to execute its business plans. The Company
is dependent on obtaining additional working capital funding from the sale of equity and/or debt securities in order to continue to execute
its development plans and continue operations. Without additional funding, there is substantial doubt about the Company’s ability
to continue as a going concern for twelve months from the date of these financial statements.
Off-Balance
Sheet Arrangements
We
have no obligations, assets or liabilities, which would be considered off-balance sheet arrangements as of June 30, 2024. We do not participate
in transactions that create relationships with unconsolidated entities or financial partnerships, often referred to as variable interest
entities, which would have been established for the purpose of facilitating off-balance sheet arrangements. We have not entered into
any off-balance sheet financing arrangements, established any special purpose entities, guaranteed any debt or commitments of other entities,
or purchased any non-financial assets.
Emerging
Growth Company Status
We
are an “emerging growth company”, as defined in the JOBS Act, and, for as long as we continue to be an emerging growth company,
we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies but not to emerging
growth companies, including, but not limited to, not being required to have our independent registered public accounting firm audit our
internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive
compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote
on executive compensation and stockholder approval of any golden parachute payments not previously approved. As an emerging growth company
we can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We intend
to avail ourselves of these options. Once adopted, we must continue to report on that basis until we no longer qualify as an emerging
growth company.
We
will cease to be an emerging growth company upon the earliest of: (i) the end of the fiscal year following the fifth anniversary of our
initial public offering; (ii) the first fiscal year after our annual gross revenue are $1.07 billion or more; (iii) the date on which
we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt securities; or (iv) the end of
any fiscal year in which the market value of our common stock held by non-affiliates exceeded $700 million as of the end of the second
quarter of that fiscal year. We cannot predict if investors will find our common stock less attractive if we choose to rely on these
exemptions. If, as a result of our decision to reduce future disclosure, investors find our common stock less attractive, there may be
a less active trading market for our common stock and the price of our common stock may be more volatile.
Critical Accounting Policies
Our
financial statements are prepared in accordance with Generally Accepted Accounting Principals (GAAP). The preparation of these financial
statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue, expenses and
related disclosures. We evaluate our estimates and assumptions on an ongoing basis. Estimates are based on historical experience and
various other assumptions that we believe to be reasonable under the circumstances. Actual results could differ from these estimates.
Our
critical accounting policies are those that materially affect its financial statements and involve difficult, subjective, or complex
judgments by management. A thorough understanding of these critical accounting policies is essential when reviewing our financial statements.
We believe that the critical accounting policies listed below involve the most difficult management decisions because they require the
use of significant estimates and assumptions as described above.
Licenses
The
Company acquires medical licenses for the treatment of medical conditions to market and sell in the future. The initial asset cost is
the cost to acquire the license. Once in use, the Company amortizes the license cost over the useful life using the straight-line method.
Patents
The
Company records the cost to acquire a patent as the initial asset cost. Once the patents are approved and in use, and assuming no litigations
expenses, the Company amortizes the patent cost over the useful life using the straight-line method. The amortization period will not
exceed the lifespan of the protection afforded by the patent. If the expected useful life of the patent is even shorter, the Company
will use the useful life for amortization purposes.
Thus,
the shorter length of a patent’s useful life and its legal life will be used for the amortization period.
Valuation
of Long-Lived and Intangible Assets
We
assess the impairment of long-lived and intangible assets periodically, or at least annually, and whenever events or changes in circumstances
indicate that the carrying value may not be recoverable. Factors considered important, which could trigger an impairment review, include
the following: significant underperformance relative to historical or projected future cash flows; significant changes in the manner
of use of the assets or the strategy of the overall business; and significant negative industry trends. When management determines that
the carrying value of long-lived and intangible assets may not be recoverable, impairment is measured as the excess of the assets’
carrying value over the estimated fair value. Management is not aware of any other impairment changes that may currently be required;
however, we cannot predict the occurrence of events that might adversely affect the reported values in the future. On an annual basis,
the Company tests the long-lived and intangible assets for impairment based on the projected net present value of cash flows for each
asset. Prior to the annual impairment test, if circumstances change and a long-lived or intangible asset is deemed impaired, an impairment
loss will be immediately recognized in the statements of operations. At December 31, 2023, the date of the last impairment test, it was
determined the estimated fair value exceeded the carry value by in excess of 50%.
Research
and Development Cost
Research
and development (R&D) costs are expensed as incurred. R&D costs are related to the Company’s internally funded development
of the Company’s medical licenses and patents. The Company had R&D costs of $-0- for the three and nine months ended September
30, 2023 and 2022.
Emerging
Growth Company Status
Section 107
of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided
in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging
growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies.
Section 107 of the JOBS Act provides that any decision to opt out of the extended transition period for complying with new or revised
accounting standards is irrevocable. We have elected to use this extended transition period under the JOBS Act.
Section
16(a) Beneficial Ownership Reporting Compliance
Under
Section 16(a) of the Securities Exchange Act of 1934, as amended, an officer, director, or greater-than-10% shareholder of the Company
must file a Form 4 reporting the acquisition or disposition of Company’s equity securities with the Securities and Exchange Commission
no later than the end of the second business day after the day the transaction occurred unless certain exceptions apply. Transactions
not reported on Form 4 must be reported on Form 5 within 45 days after the end of the Company’s fiscal year. Such persons must
also file initial reports of ownership on Form 3 upon becoming an officer, director, or greater-than-10% shareholder.
Critical
Accounting Policies and Estimates
Management’s
Discussion and Analysis of its Financial Condition and Results of Operations is based upon our consolidated financial statements, which
have been prepared in accordance with accounting principles generally accepted in the United States of America GAAP. The preparation
of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities,
revenue and expenses, and related disclosure of contingent assets and liabilities. On an on- going basis, we evaluate our estimates,
including those related to the reported amounts of revenues and expenses and the valuation of our assets and contingencies. We
believe our estimates and assumptions to be reasonable under the circumstances. However, actual results could differ from those estimates
under different assumptions or conditions. Our consolidated financial statements are based on the assumption that we will continue as
a going concern. If we are unable to continue as a going concern, we would experience additional losses from the write-down of
assets.
DIRECTORS,
EXECUTIVE OFFICERS, PROMOTERS
AND
CONTROL PERSONS
The
following persons listed below have been retained to provide services as directors until the qualification and election of his successor.
All holders of common stock will have the right to vote for directors.
The
board of directors has primary responsibility for adopting and reviewing implementation of the business plan of the registrant, supervising
the development business plan, review of the officers’ performance of specific business functions. The board is responsible
for monitoring management, and from time to time, to revise the strategic and operational plans of the registrant. A director shall
be elected by the shareholders to serve until the next annual meeting of shareholders, or until his or her death, or resignation and
his or her successor is elected.
The
Executive Officer and Directors are:
| Name |
|
Age |
|
Title |
| Joseph
Sinkule |
|
70 |
|
Chairman,
Chief Executive Officer |
| Peter
Moriarty |
|
75 |
|
Chief
Operating Officer |
| Jeffrey
LeBlanc |
|
48 |
|
Chief
Financial Officer |
| Shalom
Z. Hirschman |
|
85 |
|
Director |
| Samuel
Zentman |
|
78 |
|
Director |
| Jon
W. McGarity |
|
81 |
|
Director |
Executive
Officers
Joseph
Sinkule, Pharm.D. — Dr. Sinkule has evolved from a chemist, scientist and clinical researcher into a successful
businessman and serial entrepreneur. In 2015, he founded and has served as Chief Executive Officer and Chairman of Anew Oncology, Inc.
In 2022, Dr. Sinkule became the Chairman and Chief Executive Officer of ANEW MEDICAL which acquired Anew Oncology. He received his B.S
in Chemistry from the University of Nebraska in 1976 and a doctorate in pharmacy, pharmacology and pharmacokinetics (Pharm.D.) from University
of Nebraska Medical Center in 1980. His post-doctoral training was at St. Jude Children’s Research Hospital in Memphis, TN and
he held several academic positions at the University of Chicago and the University of Michigan. He joined the pharmaceutical and biotechnology
industry in 1990 and held key management and senior directorship positions for over 15 years including Senior Vice President and
Board member of Techniclone International, a Nasdaq-listed biotech company at the age of 33 years old. He founded, was the CEO,
and served on the Board of two biopharmaceutical companies — Virionics Corporation from 2005-2008 and Apthera, Inc.
from 2008-2012. He founded and served as CEO and Chairman of the Board of Anew Oncology, Inc. in 2015, which became ANEW MEDICAL,
INC. in 2019. Joseph has been involved with drug development for the past 40 years. He has been a consultant and advisor to several
companies and academics in the fields of cell and gene therapy for the past 18 years. Dr. Sinkule is also the Chair of the
Corporate Governance Committee. Dr. Sinkule is qualified to serve on our Board of Directors due to his extensive management experience,
drug development experience, business, finance, and entrepreneurial experiences, as well as his international contacts and relationships.
Peter
Moriarty — Mr. Moriarty has extensive experience in the pharmaceutical industry both in the United States
and internationally. He was a co-founder of Shire Pharmaceuticals, an international specialty pharmaceutical company acquired by Takeda
Pharmaceuticals (TAK — NASDAQ). He was also the co-founder of Prismic Pharmaceuticals, where he was Chairman and Chief
Executive from 2013 to 2018 and then, as Executive Chairman, led the company’s acquisition by FSD Pharma (HUGE —Nasdaq)
in 2019. Since 2019, Mr. Moriarty has acted as an independent consultant to pharmaceutical companies. Peter’s earlier career spanned
management positions within Warner-Lambert/Parke-Davis and Schering-Plough, including leadership positions overseas as well as in the
United States. Additionally, he led the Sales Force Automation and Software Products Division at Walsh America (acquired by NDC),
and led Corporate Development at Ixsys/Applied Molecular Evolution acquired by Eli Lilly (LLY —NYSE). He was subsequently
the founder, Chairman and Chief Executive Officer of iPhysicianNet, Inc. and Clinical Information Network. Mr. Moriarty attended Aston
University School of Law in conjunction with his training to become a Solicitor (Attorney) under the rules and regulations of the British
Law Society 5-year Articles program. He passed the Final/Qualifying examinations in Contract Law (with distinction), Criminal Law, Tortious
Law, and English Legal System (US accredited as equivalent to a Masters’ Degree).
Jeffrey
LeBlanc — Mr. LeBlanc has over 20 years of experience in managing financial operations, investing, advising Fortune 500
companies, and launching new ventures. He is the co-founder of Winvest Acquisition Corp. (Ticker: WINV), a special purpose acquisition
company. Prior to Winvest, Mr. LeBlanc launched Out of Print, a direct-to-consumer merchandise platform that was acquired by Penguin
Random House in 2017. He previously served in investment roles at Greenlight Capital and GE Capital, and started his career at McKinsey
and Co. Mr. LeBlanc currently serves on the Board of Directors of Cactus Acquisition Corp (Ticker: CCTSU), and previously served on the
Boards of Riot New Media Group and Books For Africa. He received an MBA from Harvard Business School and a BS in Chemical Engineering
from MIT.
Non-Executive
Directors
Shalom
Hirschman, MD — Dr. Hirschman has had a long career as an academic physician, research scientist, educator,
and, most recently, biotechnology entrepreneur and consultant. Dr. Hirschman served as intern and resident in medicine at The Massachusetts
General Hospital of the Harvard Medical School. Following years of research in molecular biology and molecular virology at the National
Institutes of Health (NIH), he was recruited to the world-renowned founding faculty of The Mount Sinai School of Medicine where he was
appointed as Director of the Division of Infectious Diseases and Professor of Medicine. Dr. Hirschman served for three decades as
Director of the Division of Infectious Diseases, and, in addition, for many years as Vice-Chairman of the Department of Medicine,
at The Mount Sinai School of Medicine and The Mount Sinai Hospital. Included among Dr. Hirschman’s many contributions to medical
research during his tenure at Mt. Sinai are the discovery of the DNA polymerase of the hepatitis B virus (HBV), the characterization
of the viral DNA, the successful replication of HBV in cell culture and the first description of AIDS as an immunologic disease. Dr. Hirschman
also served as Vice-Chairman and then as Chairman of the Microbiology Section of the New York Academy of Sciences. He retired from
Mount Sinai and joined Advanced Viral Research Corp. as its Chief Executive Officer, President, and Chief Scientific Officer. There he
successfully created a new type of peptide-based drug, built a FDA approved manufacturing facility and brought this drug into clinical
trials under INDs with the US FDA. He then took the positions as Senior Vice-President for Graduate and Professional Education and
Senior Vice-President for Graduate Academic Affairs at Touro College and the Touro University System. Dr. Hirschman is one of the
three founders of Touro College and the Touro University System and served as a member of the Board of Directors for more than three
decades. He initiated and negotiated the purchase of New York Medical College by Touro College. Since retiring from Touro University.
During the past five years Dr. Hirschman has been a consultant to educational institutions, biotechnology companies and biotechnology
investment funds, especially to Sunrise Securities Corporation. Dr. Hirschman is qualified to serve as a director because of his
clinical and medical knowledge base, his international relationships, his medical guidance on clinical development aspects, and business
experience.
Samuel
Zentman, PhD — Dr. Zentman graduated from Wayne State University and the University of Michigan and holds a
Ph.D. doctorate in Complex Analysis. He was a Mathematics professor at several divisions of the University of Detroit. And worked as
a systems analyst, manager of Engineering Computer Center, and director of the Corporate Computer Center of American Motors Corporation.
Dr. Zentman was the CFO and then CEO of Manhattan Textile Corporation, a privately held export firm in New York city. He was
a Board member of several Tech and Medical start-ups including: Neuromedical Systems Inc., Amplification Technologies, Inc., Power Safe
Technology Corp, and Hinson Hale Medical Technologies Inc. Sam is currently a Board member of Acorn Energy where he has serv3ed for over
the past 15 years, and is the Chairman of the Audit Committee, and member of the Nominating Committee and Compensation Committee.
Sam has extensive experience in dealing with early stage medical and technology companies. He also serves as the Chairman of the Board
of several national non-profits organizations devoted to the quality of education in the U.S. and abroad. We believe Dr. Zentman
is qualified to serve on our Board because of his decades of expertise in business management, public and private company finance, his
insight in medical technologies, and his global contacts and business relationships.
Jon
W. McGarity — Mr. McGarity is the President & CEO of EthiX Associates, which he founded in February 1996
and is a consultancy business serving the business needs of the healthcare industry with a focus on pharmaceuticals and biotechnology.
In addition, he has been the Chief Operating Officer of MiClimate, Inc. since 2022 which has developed the first body temperature regulation
device resulting in a change in the way people manage individual temperature sensitivities. He co-founded NeuroEM Therapeutics, Inc.,
which is currently evaluating transcranial electromagnetic therapy (TEMT) for the treatment of cognitive functioning in Alzheimer’s
disease patients, and has been its Chief Business Officer since 2021. In addition he currently is an Advisor to the Biodesign Institute
at Arizona State University. He is an original and current member of the Arizona Biosciences Roadmap Committee which provides strategic
direction to the biosciences industry in Arizona. His pharmaceutical experience includes senior management positions with GlaxoSmithKline,
Bristol Myers Squibb and Novartis (Sandoz Pharmaceuticals). Mr. McGarity has launched over 40 products as well as completing numerous
business development deals involving product acquisitions, licensing, co-marketing and promotional arrangements. Mr. McGarity’s
experience having held executive positions at “big pharma” companies, start-up operations, drug and biologics regulations,
marketing and sales, and finance makes him qualified to serve as a director.
Significant
Employees
We
have no significant employees who are not executive officers.
Family
Relationships
No
officer or director of the registrant has a family relationship with any other member of the registrant.
Involvement
in Certain Legal Proceedings
None
of our directors, executive officers, significant employees, promoters or control persons has been involved in any legal proceeding in
the past 10 years that would require disclosure under Item 401(f) of Regulation S-K promulgated under the Securities Act.
Promoters
and Control Persons
Not
applicable.
Code
of Ethics
The
Company adopted a written code of ethics that applies to the Company’s principal executive officer, principal financial officer,
principal accounting officer and controller and any persons performing similar functions.
Corporate
Governance
Committees
of the Board of Directors
The
board of directors has an audit committee, a compensation committee, a nominating and corporate governance committee and an executive
committee, Each of which will have the composition and responsibilities described below upon completion of the business combination.
Members will serve on these committees until their resignation or until otherwise determined by the board of directors.
Audit
Committee
The
audit committee consists of Samuel Zentman and Jon McGarity with Samuel Zentman serving as chairperson. Each of Samuel Zentman and Jon
McGarity and satisfy the requirements for independence and financial literacy under the rules and regulations of Nasdaq and the SEC and
Samuel Zentman qualifies as an “audit committee financial expert” as defined in the SEC rules and regulations and satisfies
the financial sophistication requirements of Nasdaq. The audit committee is responsible for, among other things:
| ● | selecting
and hiring the Company’s registered public accounting firm; |
| ● | evaluating
the performance and independence of the Company’s registered public accounting firm; |
| ● | approving
the audit and pre-approving any non-audit services to be performed by the Company’s
registered public accounting firm; |
| ● | reviewing
the integrity of the Company’s financial statements and related disclosures and reviewing
the Company’s critical accounting policies and practices; |
| ● | reviewing
the adequacy and effectiveness of the Company’s internal control policies and procedures
and the Company’s disclosure controls and procedures; |
| ● | overseeing
procedures for the treatment of complaints relating to accounting, internal accounting controls
or audit matters; |
| ● | reviewing
and discussing with management and the registered public accounting firm the results of the
annual audit, the Company’s quarterly financial statements and the Company’s
publicly filed reports; |
| ● | establishing
procedures for employees to anonymously submit concerns about questionable accounting or
audit matters; |
| ● | reviewing
and approving in advance any proposed related-person transactions; and |
| ● | preparing
the audit committee report that the SEC requires in the Company’s annual proxy statement. |
Compensation
Committee
The
compensation committee consists of Jon McGarity, Samuel Zentman and Shalom Hirschman, with Jon McGarity serving as chairperson. Each
of Jon McGarity, Samuel Zentman and Shalom Hirschman satisfies the requirements for independence under the rules and regulations of Nasdaq
and the SEC. The compensation committee of Public ANEW will be responsible for, among other things:
| ● | determining,
or recommending to the board of directors for determination, the compensation of the Company’s
executive officers, including the chief executive officer; |
| ● | overseeing
and setting compensation for the members of the board of directors; |
| ● | administering
the Company’s equity compensation plans; |
| ● | Overseeing
the Company’s overall compensation policies and practices, compensation plans, and
benefits programs; and |
| ● | preparing
the compensation committee report that the SEC will require in the Company’s annual
proxy statement. |
Nominating
and Corporate Governance Committee
The
nominating and corporate governance committee consists of Shalom Hirschman, Jon McGarity and Samuel Zentman, with Shalom Hirschman serving
as chairperson. Each of Shalom Hirschman, Jon McGarity and Samuel Zentman satisfies the requirements for independence under the rules
and regulations of Nasdaq and the SEC. The nominating and corporate governance committee will be responsible for, among other things:
| ● | evaluating
and making recommendations regarding the composition, organization and governance of the
board of directors and its committees; |
| ● | reviewing
and making recommendations with regard to the Company’s corporate governance guidelines
and compliance with laws and regulations; |
| ● | reviewing
conflicts of interest of the Company’s directors and officers and proposed waivers
of the Company’s corporate governance guidelines and code of business conducts and
ethics; and |
| ● | evaluating
the performance of the board of directors and its committees. |
Executive
Compensation
The
following tables sets forth the compensation paid to officers and board of directors during the fiscal years ended December 31, 2022
and 2023
2022
Compensation
The
following table provides information regarding the executive compensation earned by, or paid to, the executive officers of the Company
during 2022.
| Name and Principal Position | |
Salary
($) | | |
Bonus
($) | | |
Equity
Awards
($) | | |
Total
($) | |
Joseph
Sinkule
Chief Executive Officer | |
$ | 240,000 | | |
| — | | |
| — | | |
$ | 240,000 | |
Shalom
Hirschman
Medical Advisor and Director | |
$ | 65,000 | | |
| — | | |
| — | | |
$ | 65,000 | |
Peter
Moriarty
Chief Operating Officer |
|
$ | 10,000 | | |
| — | | |
| — | | |
$ | 10,000 | |
2023
Compensation
The
following table provides information regarding the executive compensation earned by, or paid to, the executive officers of the Company
during 2023.
| Name and Principal Position | |
Salary
($) | | |
Bonus
($) | | |
Equity
Awards
($) | | |
Total
($) | |
Joseph
Sinkule
Chief Executive Officer | |
$ | 160,000 | | |
| — | | |
| — | | |
$ | 160,000 | |
Shalom
Hirschman
Director and Medical Advisor | |
$ | 45,000 | | |
| | | |
| | | |
| | |
Limitations
of Director Liability and Indemnification of Directors, Officers and Employees
Our
Certificate of Incorporation limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law provides
that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors.
Our
amended and restated Bylaws provide that we will indemnify our directors and officers to the fullest extent permitted by law and may
indemnify employees and other agents. Our amended and restated Bylaws also provide that we are obligated to advance expenses incurred
by a director or officer in advance of the final disposition of any action or proceeding.
We
also maintain customary directors’ and officers’ liability insurance.
Our
amended and restated Bylaws, subject to the provisions of Delaware Law, contain provisions which allow the corporation to indemnify any
person against liabilities and other expenses incurred as the result of defending or administering any pending or anticipated legal issue
in connection with service to us if it is determined that person acted in good faith and in a manner which he or she reasonably believed
was in the best interest of the corporation. Insofar as indemnification for liabilities arising under the Securities Act of 1933 as amended,
or the Securities Act, may be permitted to our directors, officers and controlling persons, we have been advised that in the opinion
of the Securities and Exchange Commission, such indemnification is against public policy as expressed in the Securities Act and is, therefore,
unenforceable.
The
limitation of liability and indemnification provisions in our amended and restated Bylaws may discourage stockholders from bringing a
lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against
directors and officers, even though an action, if successful, might provide a benefit to us and our stockholders. Our results of operations
and financial condition may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant
to these indemnification provisions.
At
present, there is no pending litigation or proceeding involving any of our directors or officers as to which indemnification is required
or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS
AND
MANAGEMENT
The
table below sets forth the beneficial ownership of our common stock, as of August 30, 2024, by:
| |
● |
each
person who is the beneficial owner of more than 5% of the Company’s common stock; |
| |
● |
each
of the Company’s current executive officers and directors; and |
| |
● |
all
executive officers and directors of the Company, as a group. |
Beneficial
ownership is determined according to the rules of the SEC, which generally provide that a person has beneficial ownership of a security
if he, she or it possesses sole or shared voting or investment power over that security or the right to acquire such power within 60
days.
The
beneficial ownership is based on 21,263,515 shares of the Company’s Common Stock issued and outstanding.
Unless
otherwise indicated, the Company believes that all persons named in the table have sole voting and investment power with respect to all
Common Stock beneficially owned by them.
The
business address of each of the directors and officers will be 13576 Walnut Street, Suite A, Omaha, NE 68144.
| Name
and Address of Beneficial Owner(1) | |
Number of
Shares | | |
Percent
Owned | |
| Directors and Executive Officers | |
| | |
| |
| Joseph Sinkule | |
| 3,514,932 | | |
| 16.5 | % |
| Peter Moriarty | |
| 200,000 | | |
| 0.9 | % |
| Jeff LeBlanc | |
| - | | |
| = | |
| Shalom Z. Hirschman | |
| 400,000 | | |
| 1.9 | % |
| Samuel Zentman | |
| 375,000 | | |
| 1.8 | % |
| Jon W. McGarity | |
| 30,000 | | |
| * | |
| | |
| | | |
| | |
| All directors and executive officers as a group
(6 individuals) | |
| 4,519,932 | | |
| 21.3 | % |
| | |
| | | |
| | |
| Five Percent Holders: | |
| | | |
| | |
| Redwoods Capital, LLC (2) | |
| 3,478,667 | | |
| 16.4 | % |
| Chardan Capital Markets, LLC
(3) | |
| 1,740,000 | | |
| 8.2 | % |
| Upper Clapton, LLC | |
| 2,773,925 | | |
| 13.0 | % |
| Centaurus Investment Group Ltd | |
| 1,610,000 | | |
| 7.6 | % |
| All directors, executive officers and five
percent holders as a group 10 individuals) | |
| 14,122,524 | | |
| 66.4 | % |
| * |
Less
than 1%. |
| (1) |
Based
on a total of 21,263,515 shares of common stock issued and outstanding as of August 30, 2024 |
| (2) |
Redwoods
Capital LLC, a Delaware limited liability company controlled by Min Gan.
Includes 415,000 shares of common stock issuable upon the exercise
of a warrant |
| (3) |
Includes
115,000 shares of common stock issuable upon the exercise of a warrant but excludes (i) 345,000 shares of Common Stock included in
the unit issuable upon exercise of the underwriter’s purchase option (the “UPO”) held by Chardan, (ii) 115,000
shares of Common Stock issuable upon exercise of the warrant held by Chardan being registered herein and (iii) 345,000 shares of
Common Stock issuable upon exercise of the warrant (the “UPO Warrants”) included in the unit issuable upon exercise of
UPO. |
Changes
in Control
There
are no present arrangements or pledges of our securities that may result in a change in control of the registrant.
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS
Transactions
with Related Persons
On
October 10, 2021, our wholly-owned subsidiary ANEW Medical, Inc., a Wyoming corporation, and Dr. Joseph Sinkule entered into an Employment
Agreement. Pursuant to the Employment Agreement, Dr. Sinkule receives an annual base salary of $240,000 payable which shall increase
to $360,000 in the event that the Company raises a total of $5,000,000 or more in equity and/or debt financing. The term of the agreement
terminates on October 9, 2024.
On
August 15, 2024, in connection with his appointment as the Company’s Chief Operating Officer, on August 15, 2024, Mr. Moriarty
and the Company entered an Employment Agreement for a term of three years. Pursuant to the Employment Agreement, Mr. Moriarty will receive
an annual base salary of $300,000, and will receive an initial equity award of shares of the Company’s common stock of 100,000
shares and an additional equity award of 400,000 shares of the Company’s common stock, with 200,000 of such shares vesting on the
first anniversary of the agreement and 200,000 of such shares vesting on the second anniversary of the agreement. In addition, Mr. Moriarty
will be eligible to participate in the Company’s annual bonus program for executives.
On
August 15, 2024, in connection with his appointment as the Company’s Chief Financial Officer, Mr. LeBlanc and the Company entered
an Employment Agreement for a term of three years. Pursuant to the Employment Agreement, Mr. LeBlanc will receive an annual base salary
of $325,000, and will receive an initial equity award of shares of the Company’s common stock of 100,000 shares and an additional
equity award of 400,000 shares of the Company’s common stock, with 200,000 of such shares vesting on the first anniversary of the
agreement and 200,000 of such shares vesting on the second anniversary of the agreement. In addition, Mr. LeBlanc will be eligible to
participate in the Company’s annual bonus program for executives.
Promoters
and Certain Control Persons
Except
as indicated under the heading “Transactions with Related Persons” above, there have been no transactions since the beginning
of our last fiscal year, or any currently proposed transaction in which we were or are to be a participant and the amount involved exceeds
the lesser of $120,000 or one percent of the average of our total assets at year-end for the last three completed fiscal years.
DESCRIPTION
OF CAPITAL STOCK
The
following statements constitute brief summaries of our articles of incorporation and bylaws.
Authorized
Capital
General
Our
second amended and restated certificate of incorporation authorizes the issuance of 1,000,000,000 shares of common stock, par value $0.0001,
and 100,000,000 shares of undesignated preferred stock, $0.0001 par value. As of the date of this Registration Statement, 22,163,515
shares of common stock are issued and outstanding and no preferred shares are issued or outstanding. The following description summarizes
all of the material terms of our securities. Because it is only a summary, it may not contain all the information that is important to
you. For a complete description you should refer to our second amended and restated certificate of incorporation and bylaws, and the
forms of warrant agreement, which are filed as exhibits to this prospectus.
Common
Stock
Our
common stock has the following powers, rights, qualifications, limitations and restrictions:
| 1. | The
holders of the common stock shall be entitled to one vote for each share of common stock
held by them of record at the time for determining the holders thereof entitled to vote. |
| 2. | In
the event of a voluntary or involuntary liquidation, distribution or sale of assets, dissolution
or winding up of the registrant, the holders of the common stock shall be entitled to receive
all of the remaining assets of the registrant, tangible and intangible, of whatever kind
available for distribution to stockholders, ratably in proportion to the number of common
shares held by each. |
Preferred
Stock
No
shares of preferred stock are issued or outstanding. Our board of directors is empowered, without stockholder approval, to issue preferred
stock with dividend, liquidation, conversion, voting or other rights which could adversely affect the voting power or other rights of
the holders of common stock. However, the underwriting agreement prohibits us, prior to a business combination, from issuing preferred
stock which participates in any manner in the proceeds of the trust account, or which votes as a class with the common stock on our initial
business combination. We may issue some or all of the preferred stock as a method of discouraging, delaying or preventing a change in
control of us. Although we do not currently intend to issue any shares of preferred stock, we reserve the right to do so in the future.
Warrants
Each
redeemable warrant entitles the holder thereof to purchase one share of common stock at a price of $11.50 per share, subject to adjustment
as discussed below. The warrants became exercisable upon the completion of our business combination on June 21, 2024. However, no warrants
will be exercisable for cash unless we have an effective and current registration statement covering the issuance of the common stock
issuable upon exercise of the warrants and a current prospectus relating to such common stock. Notwithstanding the foregoing, if a registration
statement covering the issuance of the common stock issuable upon exercise of the warrants is not effective within 90 days from the closing
of our business combination, warrant holders may, until such time as there is an effective registration statement and during any period
when we shall have failed to maintain an effective registration statement, exercise warrants on a cashless basis pursuant to an available
exemption from registration under the Securities Act. If an exemption from registration is not available, holders will not be able to
exercise their warrants on a cashless basis. The warrants will expire five years from the closing of our business combination at 5:00
p.m., New York City time or earlier upon redemption or liquidation.
We
may redeem the outstanding warrants:
| ● | in
whole and not in part; |
| ● | at
a price of $0.01 per warrant; |
| ● | upon
a minimum of 30 days’ prior written notice of redemption, which we refer to as the
30-day redemption period; and |
| ● | if,
and only if, the last reported sale price of our common stock equals or exceeds $16.50 per
share (as adjusted for stock splits, stock dividends, reorganizations, recapitalizations
and the like) for any 20 trading days within a 30-trading day period ending on the third
trading day prior to the date on which we send the notice of redemption to the warrant holders. |
The
right to exercise will be forfeited unless the warrants are exercised prior to the date specified in the notice of redemption. On and
after the redemption date, a record holder of a warrant will have no further rights except to receive the redemption price for such holder’s
warrant upon surrender of such warrant.
The
redemption criteria for our warrants have been established at a price which is intended to provide warrant holders a reasonable premium
to the initial exercise price and provide a sufficient differential between the then-prevailing share price and the warrant exercise
price so that if the share price declines as a result of our redemption call, the redemption will not cause the share price to drop below
the exercise price of the warrants.
We
will not redeem the warrants unless a registration statement under the Securities Act covering the shares of common stock issuable upon
exercise of the warrants is effective and a current prospectus relating to those shares of common stock is available throughout the 30-day
redemption period, except if the warrants may be exercised on a cashless basis and such cashless exercise is exempt from registration
under the Securities Act. If and when the warrants become redeemable by us, we may not exercise our redemption right if the issuance
of shares of common stock upon exercise of the warrants is not exempt from registration or qualification under applicable state blue
sky laws or we are unable to effect such registration or qualification.
If
we call the warrants for redemption as described above, our management will have the option to require all holders that wish to exercise
warrants to do so on a “cashless basis.” In such event, each holder would pay the exercise price by surrendering the warrants
for that number of shares of common stock equal to the quotient obtained by dividing (x) the product of the number of shares of common
stock underlying the warrants, multiplied by the difference between the exercise price of the warrants and the fair market value by (y)
the fair market value. The “fair market value” for this purpose shall mean the average reported last sale price of the common
stock for the 10 trading days ending on the third trading day prior to the date on which the notice of redemption is sent to the holders
of warrants. Whether we will exercise our option to require all holders to exercise their warrants on a “cashless basis”
will depend on a variety of factors including the price of our common stock at the time the warrants are called for redemption, our cash
needs at such time and concerns regarding dilutive share issuances.
The
warrants are issued in registered form under a warrant agreement between Continental Stock Transfer & Trust Company, as warrant agent,
and us. The warrant agreement provides that the terms of the warrants may be amended without the consent of any holder to cure any ambiguity
or correct any defective provision, but requires the approval, by written consent or vote, of the holders of a majority of the then outstanding
warrants (including the private warrants) in order to make any change that adversely affects the interests of the registered holders.
The
exercise price and number of common stock issuable on exercise of the warrants may be adjusted in certain circumstances including in
the event of a share dividend, extraordinary dividend or our recapitalization, reorganization, merger or consolidation. However, the
warrants will not be adjusted for issuances of common stock at a price below their respective exercise prices.
The
warrants may be exercised upon surrender of the warrant certificate on or prior to the expiration date at the offices of the warrant
agent, with the exercise form on the reverse side of the warrant certificate completed and executed as indicated, accompanied by full
payment of the exercise price (or on a cashless basis, if applicable), by certified or official bank check payable to us, for the number
of warrants being exercised. The warrant holders do not have the rights or privileges of holders of common stock and any voting rights
until they exercise their warrants and receive common stock. After the issuance of common stock upon exercise of the warrants, each holder
will be entitled to one vote for each share held of record on all matters to be voted on by stockholders.
Except
as described above, no warrants will be exercisable and we will not be obligated to issue common stock unless at the time a holder seeks
to exercise such warrant, a prospectus relating to the common stock issuable upon exercise of the warrants is current and the common
stock have been registered or qualified or deemed to be exempt under the securities laws of the state of residence of the holder of the
warrants. Under the terms of the warrant agreement, we have agreed to use our best efforts to meet these conditions and to maintain a
current prospectus relating to the common stock issuable upon exercise of the warrants until the expiration of the warrants. However,
we cannot assure you that we will be able to do so and, if we do not maintain a current prospectus relating to the common stock issuable
upon exercise of the warrants, holders will be unable to exercise their warrants and we will not be required to settle any such warrant
exercise. If the prospectus relating to the common stock issuable upon the exercise of the warrants is not current or if the common stock
is not qualified or exempt from qualification in the jurisdictions in which the holders of the warrants reside, we will not be required
to net cash settle or cash settle the warrant exercise, the warrants may have no value, the market for the warrants may be limited and
the warrants may expire worthless.
We
have agreed that, subject to applicable law, any action, proceeding or claim against us arising out of or relating in any way to the
warrant agreement will be brought and enforced in the courts of the State of New York or the United States District Court for the Southern
District of New York.
Equity
Incentive Plan
The
Company’s Board adopted, and our have stockholders approved, our 2024 Equity Incentive Plan (referred to herein as the Equity Incentive
Plan). Although the Company does not have a formal policy with respect to the grant of equity incentive awards to the Company’s
executive officers, the Company believes that equity awards provide the Company’s executive officers with a strong link to the
Company’s long-term performance, create an ownership culture and help to align the interests of the Company’s executives
and Public the Company’s stockholders. In addition, the Company believes that equity awards with a time-based vesting feature promote
executive retention because this feature incentivizes the Company’s executive officers to remain in the Company’s employment
during the applicable vesting period. Accordingly, the Company’s board of directors periodically reviews the equity incentive compensation
of the Company’s named executive officers (“NEOs”) and from time to time may grant equity incentive awards to them.
No stock options or other equity awards were granted to the Company’s NEOs during the fiscal year ended December 31, 2023.
Outstanding
Equity Awards at Fiscal Year-End
There
were no outstanding equity awards held by our NEOs as of December 31, 2023.
Transfer
Agent
The
transfer agent for our shares of common stock and warrants is Continental Stock Transfer & Trust Company, 1 State Street, 30th Floor,
New York, NY 10004-1561.
Anti-Takeover
Provisions of Provisions of Delaware Law and Charter Documents
Our
Second Amended Certificate of Incorporation (“Certificate of Incorporation”) and amended and restated bylaws (“Bylaws”)
contain provisions that might have an anti-takeover effect. These provisions, which are summarized below, may have the effect of delaying,
deterring or preventing a change in control of our Company. They could also impede a transaction in which our stockholders might receive
a premium over the then-current market price of our common stock and our stockholders’ ability to approve transactions that they
consider to be in their best interests.
Certificate
of Incorporation. Our authorized but unissued shares of common stock and preferred stock are available for our Board to issue without
stockholder approval. We may use these additional shares for a variety of corporate purposes, including future public or private offerings
to raise additional capital, corporate acquisitions and employee benefit plans. The existence of our authorized but unissued shares of
common stock and preferred stock could render more difficult or discourage an attempt to obtain control of our Company by means of a
proxy contest, tender offer, merger or other transaction. Our authorized but unissued shares may be used to delay, defer or prevent a
tender offer or takeover attempt that a stockholder might consider in its best interest, including those attempts that might result in
a premium over the market price for the shares held by our stockholders.
Bylaws.
Certain provisions of our Bylaws may be considered to have anti-takeover effects, including advance notice requirements for director
nominations and other stockholder proposals. Our Bylaws establish advance notice procedures for stockholder proposals to be brought before
an annual meeting of stockholders, and for proposed nominations of candidates for election to our Board at an annual or special meeting
of stockholders. Generally, such notices must be received by our corporate secretary at our principal executive offices, in the case
of an annual meeting, between 90 days and 120 days prior to the first anniversary of the preceding year’s annual meeting and, in
the case of a special meeting called for the purpose of electing directors, between 90 and 120 days prior to the date of the special
meeting or within 10 days after the day on which public announcement of the date of the special meeting is first made by us. In addition,
our Board has the authority to amend or repeal our Bylaws, or to adopt new bylaws, which could have the effect of delaying, deterring
or preventing a change of control.
SELLING
STOCKHOLDERS
The
following table details the name of the Selling Stockholders, the number of shares beneficially owned by each Selling Stockholder, and
the number of shares that may be offered by each Selling Stockholder for resale under this prospectus. The Selling Stockholders may sell
up to 10,027,925 shares of common stock from time to time in one or more offerings under this prospectus. Because the Selling Stockholders
may offer all, some or none of the shares they hold, and because, based upon information provided to us, there are currently no agreements,
arrangements, or understandings with respect to the sale of any of the shares, no definitive estimate as to the number of shares that
will be held by the Selling Stockholders after the offering can be provided. The Selling Stockholders have informed us that they are
not registered broker-dealers and do not have any written or oral agreement or understanding, directly or indirectly, with any person
to distribute the securities. Furthermore, the Selling Stockholders are not an affiliate of a broker-dealer. The following table has
been prepared on the assumption that all shares offered under this prospectus will be sold to parties unaffiliated with the Selling Stockholders.
Except for the ownership of shares of common stock and/or the Warrants, the Selling Stockholders have not had any material relationship
with us within the past three years.
In
the event of stock splits, stock dividends or similar transactions involving the shares of common stock, the number of shares of common
stock registered shall, unless otherwise expressly provided, automatically be deemed to cover the additional securities to be offered
or issued pursuant to Rule 416 promulgated under the Securities Act of 1933, as amended (the “Securities Act”). In
the event that the adjustment provisions of the securities purchase agreement require the registrant to issue more shares than are being
registered in this registration statement, for reasons other than those stated in Rule 416 of the Securities Act, the registrant will
file a new registration statement to register those additional shares.
The
table below lists the Selling Stockholders and other information regarding the beneficial ownership of the shares of common stock by
each of the Selling Stockholders. The second column lists the number of shares of common stock beneficially owned by each Selling Stockholder,
based on its ownership of the shares of common stock and warrants, as of August 30, 2024, assuming exercise of the warrants held by the
applicable Selling Stockholder on that date, without regard to any limitations on exercises.
The
third column lists the shares of common stock being offered by the Selling Stockholders pursuant to this prospectus. The fourth column
assumes the sale of all of the shares of common stock being offered by the Selling Stockholders pursuant to this prospectus.
This
prospectus generally covers the resale of the maximum number of shares of common stock issuable upon exercise of the related Warrants,
determined as if the outstanding Warrants were exercised in full as of the trading day immediately preceding the date this registration
statement was initially filed with the SEC, each as of the trading day immediately preceding the applicable date of determination and
all subject to adjustment as provided in the registration right agreement, without regard to any limitations on the exercise of the Warrants.
The fourth column assumes the sale of all of the shares offered by the Selling Stockholders pursuant to this prospectus.
| Name | |
Number
of
Shares of
Common
Stock
Owned
Prior to
Offering | | |
Maximum
Number of
Shares of
Common
Stock to be
Sold
Pursuant to
this
Prospectus* | | |
Number
of
Shares of
Common
Stock
Owned
After
Offering
(if Sold) (1)(2) | | |
%
of
Shares of
Common
Stock
Owned
After
Offering (1)(2)(3) | |
| Joseph A. Sinkule | |
| 3,514,932 | | |
| 3,500,000 | | |
| 14,932 | | |
| 0.1 | % |
| Upper Clapton, LLC | |
| 2,773,925 | | |
| 2,773,925 | | |
| 0 | | |
| 0.0 | % |
| Redwoods Capital, LLC* (5) | |
| 3,478,667 | | |
| 415,000 | | |
| 3,063,667 | | |
| 14.4 | % |
| Centaurus Investment Group Ltd | |
| 1,610,000 | | |
| 1,610,000 | | |
| 0 | | |
| 0.0 | % |
| Full and Accurate Services Limited | |
| 980,000 | | |
| 840,000 | | |
| 140,000 | | |
| 0.7 | % |
| Edward Wang | |
| 300,000 | | |
| 300,000 | | |
| 0 | | |
| 0.0 | % |
| Shalom Hirschman | |
| 240,000 | | |
| 240,000 | | |
| 0 | | |
| 0.0 | % |
| White Lion Capital, LLC | |
| 130,000 | | |
| 130,000 | | |
| 0 | | |
| 0.0 | % |
| Chardan Capital Markets, LLC (4) | |
| 1,740,000 | | |
| 115,000 | | |
| 1,625,000 | | |
| 7.6 | % |
| Peter Moriarity | |
| 200,000 | | |
| 100,000 | | |
| 100,000 | | |
| 0.5 | % |
| Richard Allsopp | |
| 1,000 | | |
| 1,000 | | |
| 0 | | |
| 0.0 | % |
| Michio Shimabukuro | |
| 1,000 | | |
| 1,000 | | |
| 0 | | |
| 0.0 | % |
| Donald Craig Willcox | |
| 1,000 | | |
| 1,000 | | |
| 0 | | |
| 0.0 | % |
| Bradley J. Willcox | |
| 1,000 | | |
| 1,000 | | |
| 0 | | |
| 0.0 | % |
| Total
Selling Stockholders | |
| 14,971,524 | | |
| 10,027,925 | | |
| 4,943,599 | | |
| 23.2 | % |
| (1) | Beneficial
ownership is determined in accordance with SEC rules and generally includes voting or investment
power with respect to common shares. Common shares subject to options, warrants or other
convertible securities currently exercisable or convertible, or exercisable or convertible
within 60 days, are counted as outstanding for computing the percentage of the person holding
such options, warrants or other convertible securities but are not counted as outstanding
for computing the percentage of any other person. |
| (2) | The
amount and percentage of common shares that will be beneficially owned by the Selling Stockholders
after completion of the offering assume that they will sell all common shares being offered
pursuant to this prospectus. |
| (3) | Based
on 21,263,515 shares of common stock issued and outstanding as of August 30, 2024. All shares
of common stock being offered pursuant to this prospectus by the Selling Stockholders are
counted as outstanding for computing the percentage beneficial ownership of such Selling
Stockholders |
| (4) | Includes
115,000 issuable upon the exercise of a warrant but excludes (i) 345,000 shares of Common
Stock included in the unit issuable upon exercise of the underwriter’s purchase option
(the “UPO”) held by Chardan, (ii) 115,000 shares of Common Stock issuable upon
exercise of the warrant held by Chardan being registered herein and (iii) 345,000 shares
of Common Stock issuable upon exercise of the warrant (the “UPO Warrants”) included
in the unit issuable upon exercise of UPO. |
| (5) | Includes
415,000 shares of common stock issuable upon the exercise of a common stock purchase warrant. |
| * | Redwoods,
LLC, a Delaware limited liability company controlled by Min Gan. |
SHARES
ELIGIBLE FOR FUTURE SALE
Upon
the date of this prospectus, there are 21,263,515 shares of common stock outstanding, none of which may be freely traded without registration
or an applicable exemption. The shares of common stock being registered pursuant to this registration statement shall be freely
tradable upon the effective date of the registration statement until the termination of the offering, unless sold.
Any
additional common shares issued in the future but not registered with the Securities and Exchange Commission are restricted within the
meaning of Rule 144 under the Securities Act, and are subject to the resale provisions of Rule 144.
At
the present time, re-sales or distributions of such shares are provided for by the provisions of Rule 144. That rule is a so-called
“safe harbor” rule that, if complied with, should eliminate any questions as to whether or not a person selling restricted
shares has acted as an underwriter.
Rule
144(d) (1) states that if the issuer of the securities is, and has been for a period of at least 90 days immediately before the sale,
subject to the reporting requirements of section 13 or 15(d) of the Exchange Act, a minimum of six months must elapse between the later
of the date of the acquisition of the securities from the issuer, or from an affiliate of the issuer, and any resale of such securities.
Sales
under Rule 144 are also subject to notice and manner of sale requirements and to the availability of current public information and must
be made in unsolicited brokers’ transactions or to a market maker.
A
person who is not an affiliate of the registrant under the Securities Act during the three months preceding a sale and who has beneficially
owned such shares for at least six months is entitled to sell the shares under Rule 144 without regard to the volume, notice, information
and manner of sale provisions. Affiliates must comply with the restrictions and requirements of Rule 144 when transferring restricted
shares even after the six month holding period has expired and must comply with the restrictions and requirements of Rule 144 in order
to sell unrestricted shares.
No
predictions can be made of the effect, if any, that market sales of common shares or the availability of such shares for sale will have
on the market price prevailing from time to time. Nevertheless, sales of significant amounts of our common shares could adversely
affect the prevailing market price of the common shares, as well as impair our ability to raise capital through the issuance of additional
equity securities.
DISCLOSURE
OF COMMISSION POSITION ON
INDEMNIFICATION
FOR SECURITIES ACT LIABILITIES
Insofar
as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of
the registrant as provided in the foregoing provisions, or otherwise, we have been advised that in the opinion of the SEC such indemnification
is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
In
the event that a claim for indemnification against such liabilities, other than the payment by us of expenses incurred or paid by a director,
officer or controlling person of the registrant in the successful defense of any action, suit or proceeding, is asserted by such director,
officer or controlling person in connection with the securities being registered, we will, unless in the opinion of its counsel the matter
has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by
it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS
ON
ACCOUNTING AND FINANCIAL DISCLOSURE
For
accounting purposes, the Business Combination was treated as a reverse acquisition and, as such, the historical financial statements
of the accounting acquirer, ANEW Wyoming which have been audited by Yusufali & Associates, LLC (“Yusufali”), became the
historical financial statements of the Company. In a reverse acquisition, a change of accountants is presumed to have occurred unless
the same accountant audited the pre-transaction financial statements of both the legal acquirer and the accounting acquirer, and such
change is generally presumed to occur on the date the reverse acquisition is completed.
On
June 24, 2024, the audit committee of the Company’s Board approved the engagement of Yusufali as the Company’s independent
registered public accounting firm to audit the Company’s consolidated financial statements for the fiscal year ending December
31, 2024. Yusufali served as the independent registered public accounting firm of ANEW Wyoming prior to the Business Combination. On
June 24, 2024, Marcum, LLP (“Marcum”), the independent registered public accounting firm for the Company prior to the Business
Combination, was informed that it was dismissed as the auditor of the Company immediately after the Closing of the Business Combination.
The
report of Marcum on Redwoods’ financial statements as of December 31, 2023 and 2022, and for the years then ended, and the related
notes to the financial statements, did not contain an adverse opinion or a disclaimer of opinion, and were not qualified or modified
as to uncertainties, audit scope or accounting principles, except that the report contained an explanatory paragraph relating to substantial
doubt about the ability of Redwoods to continue as a going concern as described in Note 1 to the financial statements.
During
the years ended December 31, 2022 and December 31, 2023, and the subsequent period through June 27, 2024, there were no disagreements
with Marcum on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which
disagreements, if not resolved to the satisfaction of Marcum, would have caused it to make a reference to the subject matter of the disagreement
in connection with its report covering such period. In addition, no “reportable events,” as defined in Item 304(a)(1)(v)
of Regulation S-K, occurred within the period of Marcum’s engagement and the subsequent period through June 27, 2024.
MARKET
FOR COMMON EQUITY AND RELATED
STOCKHOLDER
MATTERS
Our
common stock is listed on the Nasdaq Global Market under the symbol “WENA” and our warrants are listed on the Nasdaq Global
Market under the symbol “WENAW.”
For
any market that is maintained for our common stock, the resale of “restricted securities” pursuant to Rule 144 of the Commission
by members of management or other persons may have a substantial adverse impact on any such public market.
A
minimum holding period of one year is required for resales under Rule 144 until June 21, 2025. In addition, affiliates of the Company
must comply with certain other requirements, including publicly available information concerning the Company; limitations on the volume
of “restricted securities” which can be sold in any 90-day period; the requirement of unsolicited broker’s transactions;
and the filing of a Notice of Sale on Form 144.
Sales
under Rule 144 are also subject to manner of sale provisions and notice requirements and to the availability of current public information
about the Company. Under Rule 144(k), a person who has not been one of our affiliates at any time during the three months preceding a
sale, and who has beneficially owned the shares proposed to be sold for at least six months, is entitled to sell shares without complying
with the manner of sale, volume limitation or notice provisions of Rule 144.
Holders
As of August 30, 2024, we had approximately 79
shareholders of record of our common stock.
Dividend
Policy
We
have not paid any dividends to shareholders. There are no restrictions which would limit our ability to pay dividends on common equity
or that are likely to do so in the future. The Delaware General Corporation Law, however, does prohibit us from declaring dividends where,
after giving effect to the distribution of the dividend; we would not be able to pay our debts as they become due in the usual course
of business; or our total assets would be less than the sum of the total liabilities plus the amount that would be needed to satisfy
the rights of shareholders who have preferential rights superior to those receiving the distribution.
Issuer
Purchases of Equity Securities
We
have not repurchased any shares of our common stock since inception.
EXPERTS
Our
consolidated financial statements as of December 31, 2023 and 2022 appearing in this prospectus and in the registration statement have
been audited by Yusufali & Associates, LLC. an independent registered public accounting firm and are included in reliance upon such
report given upon the authority of such firm as experts in accounting and auditing.
LEGAL
PROCEEDINGS
From
time to time, we may become involved in various lawsuits and legal proceedings that may arise in the ordinary course of business. However,
litigation is subject to inherent uncertainties and an adverse result in these or other matters may arise from time to time that may
have an adverse effect on our business, financial conditions, or operating results. We are not aware of any legal proceedings or claims
that will have, individually or in the aggregate, a material adverse effect on our business, financial condition or operating results.
LEGAL
MATTERS
The
validity of the common shares being offered hereby will be passed upon by Cyruli, Shanks & Zizmor, LLP, Attorneys At Law, New York,
New York.
WHERE
YOU CAN FIND MORE INFORMATION
At
your request, we will provide you, without charge, a copy of any document filed as exhibits in this prospectus. If you want more information,
write or call us at: (833) 931-6330.
We
have filed a registration statement on Form S-1 under the Securities Act with the SEC for the securities offered hereby. This prospectus,
which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement
or the exhibits and schedules which are part of the registration statement. For additional information about us and our securities,
we refer you to the registration statement and the accompanying exhibits and schedules. Statements contained in this prospectus regarding
the contents of any contract or any other documents to which we refer are not necessarily complete.
In
each instance, reference is made to the copy of the contract or document filed as an exhibit to the registration statement, and each
statement is qualified in all respects by that reference Copies of the registration statement and the accompanying exhibits and
schedules may be inspected without charge (and copies may be obtained at prescribed rates) at the public reference facility of the SEC
at Room 1024, 100 F Street, N.E. Washington, D.C. 20549.
You
can request copies of these documents upon payment of a duplicating fee by writing to the SEC. You may call the SEC at 1-800-SEC-0330
for further information on the operation of its public reference rooms. Our filings, including the registration statement, will also
be available to you on the Internet web site maintained by the SEC at http://www.sec.gov.
CONSOLIDATED
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index
to Consolidated Financial Statements
ANEW
MEDICAL, INC.
CONSOLIDATED
BALANCE SHEETS
| | |
June 30,
2024 | | |
December 31,
2023 | |
| | |
(Unaudited) | | |
| |
| ASSETS | |
| | |
| |
| Current assets: | |
| | |
| |
| Cash | |
$ | 845,336 | | |
$ | 2,808 | |
| Prepaid expenses | |
| 154,500 | | |
| 3,840 | |
| Total current assets | |
| 999,836 | | |
| 6,648 | |
| | |
| | | |
| | |
| Other assets: | |
| | | |
| | |
| Licenses | |
| 2,261,134 | | |
| 2,137,638 | |
| Patents | |
| 48,420 | | |
| 48,420 | |
| Total other assets | |
| 2,309,554 | | |
| 2,186,058 | |
| Total assets | |
$ | 3,309,390 | | |
$ | 2,192,706 | |
| | |
| | | |
| | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable | |
$ | 33,988 | | |
$ | 151,259 | |
| Accrued expenses | |
| 250,400 | | |
| 2,460 | |
| Related party payable | |
| 31,000 | | |
| 159,000 | |
| Notes payable | |
| - | | |
| 1,308,270 | |
| Assumed Income tax payable from Merger | |
| 568,111 | | |
| - | |
| Other liabilities | |
| 15,064 | | |
| - | |
| Total current liabilities | |
| 898,563 | | |
| 1,620,989 | |
| Convertible promissory notes | |
| 3,900,000 | | |
| - | |
| Warrant liability | |
| 62,222 | | |
| - | |
| Total liabilities | |
| 4,860,785 | | |
| 1,620,989 | |
| | |
| | | |
| | |
| Commitments and contingencies (Note 7) | |
| | | |
| | |
| | |
| | | |
| | |
| STOCKHOLDERS’ EQUITY | |
| | | |
| | |
| Preferred stock, par value $0.0001, 100,000,000 shares authorized; 0 shares issued and outstanding as of June 30, 2024 and December 31, 2023, respectively | |
| - | | |
| - | |
| Common stock, par value $0.0001, 1,000,000,000 shares authorized; 15,678,898 and 15,130,393 shares issued and outstanding as of June 30, 2024 and December 31, 2023, respectively | |
| 1,568 | | |
| 1,513 | |
| Additional paid-in capital | |
| 3,678,685 | | |
| 4,493,881 | |
| Common stock to be issued | |
| 304,462 | | |
| 0 | |
| Accumulated deficit | |
| (5,536,110 | ) | |
| (3,923,677 | ) |
| Total stockholders’ equity (deficit) | |
| (1,551,395 | ) | |
| 571,717 | |
| Total liabilities and stockholders’ equity | |
$ | 3,309,390 | | |
$ | 2,192,706 | |
See
accompanying notes to the unaudited consolidated financial statements.
ANEW
MEDICAL, INC.
CONSOLIDATED
STATEMENTS OF OPERATIONS
(Unaudited)
| | |
For the
Three Months Ended | | |
For the
Three Months Ended | | |
For the
Six Months Ended | | |
For the
Six Months Ended | |
| | |
June 30,
2024 | | |
June 30,
2023 | | |
June 30,
2024 | | |
June 30,
2023 | |
| | |
| | |
| | |
| | |
| |
| Operating expenses: | |
| | |
| | |
| | |
| |
| Professional fees | |
$ | 393,266 | | |
$ | 229,035 | | |
$ | 768,862 | | |
$ | 376,305 | |
| General and administrative | |
| 2,341 | | |
| 15,217 | | |
| 48,790 | | |
| 20,547 | |
| Total operating expenses | |
| 395,607 | | |
| 244,252 | | |
| 817,652 | | |
| 396,852 | |
| | |
| | | |
| | | |
| | | |
| | |
| Operating income (loss) | |
| (395,607 | ) | |
| (244,252 | ) | |
| (817,652 | ) | |
| (396,852 | ) |
| | |
| | | |
| | | |
| | | |
| | |
| Nonoperating income (expenses): | |
| | | |
| | | |
| | | |
| | |
| Interest expense | |
| (15,064 | ) | |
| (20,157 | ) | |
| (15,064 | ) | |
| (40,093 | ) |
| Change in fair value of warrant liability | |
| (39,697 | ) | |
| - | | |
| (39,697 | ) | |
| - | |
| Other income (expenses) | |
| (1,271 | ) | |
| 20 | | |
| (251,270 | ) | |
| 73 | |
| Total nonoperating expenses | |
| (56,032 | ) | |
| (20,137 | ) | |
| (306,031 | ) | |
| (40,020 | ) |
| | |
| | | |
| | | |
| | | |
| | |
| Net income (loss) before income taxes | |
| (451,639 | ) | |
| (264,389 | ) | |
| (1,123,683 | ) | |
| (436,872 | ) |
| Income taxes | |
| - | | |
| - | | |
| - | | |
| - | |
| Net income (loss) | |
$ | (451,639 | ) | |
$ | (264,389 | ) | |
$ | (1,123,683 | ) | |
$ | (436,872 | ) |
| | |
| | | |
| | | |
| | | |
| | |
Net income (loss) per share: Basic and Diluted | |
$ | (0.03 | ) | |
$ | (0.02 | ) | |
$ | (0.07 | ) | |
$ | (0.03 | ) |
| Weighted average common shares outstanding | |
| 15,678,898 | | |
| 15,130,393 | | |
| 15,678,898 | | |
| 15,130,393 | |
See
accompanying notes to the unaudited consolidated financial statements.
ANEW
MEDICAL, INC.
CONSOLIDATED
STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
(Unaudited)
| | |
| | |
| | |
Preferred
Stock | | |
Additional | | |
Common | | |
| | |
Total | |
| | |
Common
Stock | | |
(Series
B, C and D) | | |
Paid-in | | |
Stock | | |
Accumulated | | |
Stockholder's | |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
to
be Issued | | |
Deficit | | |
Equity
(Deficit) | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| |
| Balance,
January 1, 2024, Revised | |
| 15,130,393 | | |
$ | 1,513 | | |
| 120,000 | | |
$ | 12 | | |
$ | 4,493,881 | | |
$ | - | | |
$ | (3,923,677 | ) | |
$ | 571,729 | |
| Retroactive
application of merger | |
| 548,505 | | |
| 55 | | |
| (120,000 | ) | |
| (12 | ) | |
| (1,318,672 | ) | |
| 304,200 | | |
| - | | |
| (1,014,429 | ) |
| Adjusted
balance, beginning of period* | |
| 15,678,898 | | |
| 1,568 | | |
| - | | |
| - | | |
| 3,175,209 | | |
| 304,200 | | |
| (3,923,677 | ) | |
| (442,700 | ) |
| Public
warrants assumed from SPAC | |
| - | | |
| - | | |
| - | | |
| - | | |
| 488,750 | | |
| - | | |
| (488,750 | ) | |
| - | |
| Private
warrants assumed from SPAC | |
| - | | |
| - | | |
| - | | |
| - | | |
| (22,525 | ) | |
| - | | |
| - | | |
| (22,525 | ) |
| Share-based
compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| 37,251 | | |
| 262 | | |
| - | | |
| 37,514 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (1,123,683 | ) | |
| (1,123,683 | ) |
| Balance
at June 30, 2024 | |
| 15,678,898 | | |
$ | 1,568 | | |
| - | | |
$ | - | | |
$ | 3,678,685 | | |
$ | 304,462 | | |
$ | (5,536,110 | ) | |
$ | (1,551,395 | ) |
| | |
Common Stock | | |
Preferred Stock
(Series B, C and D) | | |
Additional
Paid-in | | |
Accumulated | | |
Total
Stockholder’s | |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
Equity (Deficit) | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| |
| Balance, January 1, 2023 as recast | |
| - | | |
$ | - | | |
| 1,405,250 | | |
$ | 475 | | |
$ | 3,419,003 | | |
$ | (3,216,219 | ) | |
$ | 203,259 | |
| Retroactive application of merger | |
| 15,130,393 | | |
| 1,513 | | |
| (1,405,250 | ) | |
| (475 | ) | |
| 1,074,878 | | |
| - | | |
| 1,075,916 | |
| Adjusted balance, beginning of period* | |
| 15,130,393 | | |
| 1,513 | | |
| - | | |
| - | | |
| 4,493,881 | | |
| (3,216,219 | ) | |
| 1,279,175 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (436,872 | ) | |
| (436,872 | ) |
| Balance at June 30, 2023, Revised | |
| 15,130,393 | | |
$ | 1,513 | | |
| - | | |
$ | - | | |
$ | 4,493,881 | | |
$ | (3,653,091 | ) | |
$ | 842,303 | |
See
accompanying notes to the unaudited consolidated financial statements.
ANEW
MEDICAL, INC.
CONSOLIDATED
STATEMENTS OF CASH FLOWS
(Unaudited)
| | |
For the
Six Months Ended | | |
For the
Six Months Ended | |
| | |
June 30,
2024 | | |
June 30,
2023 | |
| | |
| | |
| |
| CASH FLOWS FROM OPERATING ACTIVITIES: | |
| | |
| |
| Net loss | |
$ | (1,123,683 | ) | |
$ | (436,872 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | | |
| | |
| Changes in fair value of warrant liability | |
| 39,697 | | |
| - | |
| Commitment fee | |
| 250,000 | | |
| | |
| Share-based compensation | |
| 37,514 | | |
| - | |
| Changes in operating assets and liabilities: | |
| | | |
| | |
| Prepaid expenses | |
| (150,660 | ) | |
| 2,750 | |
| Accounts payable | |
| (117,271 | ) | |
| 166,307 | |
| Accrued expenses | |
| 247,940 | | |
| 2,386 | |
| Related party payable | |
| (128,000 | ) | |
| - | |
| Other Liabilities | |
| 15,064 | | |
| - | |
| Net cash used in operating activities | |
$ | (929,399 | ) | |
$ | (265,429 | ) |
| | |
| | | |
| | |
| CASH FLOWS FROM INVESTING ACTIVITIES: | |
| | | |
| | |
| Acquisition of patents | |
| - | | |
| (10,000 | ) |
| Acquisition of drug license | |
| (123,497 | ) | |
| (10,000 | ) |
| Net cash used in investing activities | |
$ | (123,497 | ) | |
$ | (20,000 | ) |
| | |
| | | |
| | |
| CASH FLOWS FROM FINANCING ACTIVITIES: | |
| | | |
| | |
| Proceeds from convertible promissory note | |
| 950,000 | | |
| - | |
| Proceeds from sales of stocks and warrants, net | |
| 175,000 | | |
| - | |
| Merger proceeds net of transaction cost | |
| 770,424 | | |
| - | |
| Repayment of advance to shareholder | |
| - | | |
| 250,000 | |
| Net cash provided by financing activities | |
$ | 1,895,424 | | |
$ | 250,000 | |
| | |
| | | |
| | |
| NET CHANGE IN CASH | |
| 842,528 | | |
| (35,429 | ) |
| Cash - Beginning of period | |
| 2,808 | | |
| 75,872 | |
| Cash - End of period | |
$ | 845,336 | | |
$ | 40,443 | |
| | |
| | | |
| | |
| SUPPLEMENTAL NON-CASH FINANCING AND INVESTING ACTIVITIES: | |
| | | |
| | |
| Note payable settled with issuance of common stock | |
$ | 1,308,270 | | |
$ | - | |
| Non-cash directors and officers insurance | |
$ | 154,500 | | |
$ | - | |
| Non-cash PIPE Funds used for merger transaction close | |
$ | 2,950,000 | | |
$ | - | |
| Commitment fee paid in stock | |
$ | 250,000 | | |
$ | - | |
| Assumed income tax payable from merger | |
$ | 568,111 | | |
$ | - | |
| Assumed warrant liability from merger | |
$ | 22,525 | | |
$ | - | |
| | |
| | | |
| | |
| SUPPLEMENTAL CASH FLOW INFORMATION: | |
| | | |
| | |
| Interest Paid | |
$ | 2,460 | | |
$ | 37,707 | |
| Taxes Paid | |
$ | - | | |
$ | - | |
See
accompanying notes to the unaudited consolidated financial statements.
ANEW
MEDICAL, INC.
NOTES
TO THE UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 — ORGANIZATION
AND BUSINESS DESCRIPTION
ANEW
Medical, Inc. (“The Company” or “Public ANEW”) develops essential medicines for the treatment of chronic diseases
– cancer, cardiovascular, and neurodegenerative disorders. The Company currently has acquired two licensed platforms: a generic
drug portfolio and a biosimilar biologics platform that uses biologic therapies to treat cancer, and two proprietary, patented technologies
involving the melanocortin receptor-binding molecules and a gene therapy platform which uses a gene therapy approach to introduce a therapeutic
protein called “Klotho” inside the body to treat neurodegenerative diseases.
On
September 12, 2022, the Company acquired five market-approved anti-cancer drugs approved for sale in Germany. The Market Authorizations
(MA’s) are for four of the drugs that comprise the “FOLFOX” and “FOLFIRI” multi-drug regimens used in treatment
of metastatic colorectal and gastric cancer and in two of the drugs that are used to treat metastatic lung cancer. The drugs are important
in the treatment of many solid tumors in both childhood and adult cancers. Previously, the Company acquired two off-patent bio generic
antibodies from Reliance Life Sciences (RLS), the life science arm of Reliance Industries Pvt Ltd. of Navi Mumbai, India.
During
January 2023, the Company acquired a treatment for small drug molecules that bind to the melanocortin receptors on human cells and affect
skin pigmentation.
Business
Combinations
As
of May 30, 2023, Redwoods Acquisition Corp., a Delaware corporation and a special purpose acquisition company (“Redwoods”),
Anew Medical Sub, Inc., a Wyoming corporation (“Merger Sub”) and ANEW Medical, Inc., a Wyoming corporation (“ANEW”)
entered into a Business Combination Agreement, which was amended as of November 4, 2023 (the “Business Combination Agreement”).
On June 21, 2024 (the “Closing Date”), Merger Sub merged with and into ANEW, with ANEW continuing as the surviving corporation
and as a wholly owned subsidiary of Redwoods (the “Business Combination”). In connection with the Business Combination, on
June 21, 2024, Public ANEW (“the Company”) filed the Amended Charter with the Delaware Secretary of State, and adopted the
amended and restated bylaws (the “Amended and Restated Bylaws”), which replaced Redwoods’ Charter and Bylaws in effect
as of such time. In connection with the closing of the Business Combination (the “Closing”), Redwoods changed its name to
“ANEW Medical, Inc.” (“Public ANEW”).
For
accounting purposes, the transactions contemplated by the Business Combination are treated as a reverse acquisition and, as such, the
historical financial statements of the accounting acquirer ANEW will become the historical financial statements of Public ANEW. Under
this method of accounting, Redwoods was treated as the acquired company for financial reporting purposes. Accordingly, for
accounting purposes, the Merger was treated as the equivalent of the Company issuing shares for the net assets of Redwoods,
accompanied by a recapitalization. The net assets of Redwoods were stated at historical cost with no goodwill or other intangible
assets recorded.
Recapitalization
In
connection with the merger, Redwoods issued six million shares in exchange for all of the outstanding shares of the Company. At
$10 per Redwood’s share, the valuation of the Company was $60 million.
Immediately
after giving effect to the Business Combination, 15,130,393 shares of Company Common Stock were outstanding, from which 2,875,000 remain
in escrow for the Redwoods founders. In addition, the new Public ANEW assumed 12,030,000 warrants from Redwoods in connection with the
merger immediately exercisable and composed of 11,500,000 public warrants and 530,000 private warrants. Following the Closing, on June
21, 2024, the ANEW Common Stock and Public ANEW Warrants began trading on the Nasdaq under the symbols “WENA” and “WENAW,”
respectively. The Public Units of Redwoods automatically separated into the component securities upon consummation of the Business Combination
and, as a result, no longer trade as a separate security. Further, upon closing of the Business Combination on June 21, 2024, ANEW Medical
received approximately $181,339 in net cash proceeds. The Company assumed from Redwoods approximately $589,081 in cash.
At
Closing, pursuant to the terms of the Merger Agreement and after giving effect to the redemptions of shares of Redwoods Common Stock:
| ● | The total consideration paid at Closing (the “Merger Consideration”) by Redwoods to ANEW Medical, Inc. security holders was 6,000,000 shares of the Company common stock valued at $60 million (the “Consideration Shares”), based on an implied ANEW equity value of $60,000,000 valued at $10 per share; |
| ● | Each
share of ANEW Medical Common Stock, if any, that was owned by Redwoods, Merger Sub, ANEW
Medical, Inc. or any other affiliate of Redwoods immediately prior to the effective time
of the Merger (the “Effective Time”) was automatically cancelled and retired
without any conversion or consideration; |
| ● | Each share of Merger Sub common stock, par value $0.0001 per share (“Merger Sub Common Stock”), issued and outstanding immediately prior to the Effective Time was converted into one newly issued share of Common Stock of the Surviving Corporation. |
On March 4, 2024, in
connection with the Merger, Public ANEW entered into a convertible promissory note and Securities Purchase Agreement (“SPA”)
with certain accredited investors (the “Redwoods PIPE Investors”) for an aggregate purchase price of up to $2,000,000
(the “Redwoods PIPE Financing”), which included 750,000 bonus shares of common stock. Upon the closing of the Redwoods
PIPE Financing (funded and closed in connection with the closing of the Merger on June 21, 2024), which totaled $1,950,000,
of which $1,768,661 was used by the Company to settle transaction costs. The Company received approximately $181,339 in net cash proceeds.
On April 22, 2024, in connection with the Merger,
Public ANEW entered convertible promissory note and Securities Purchase Agreement (“SPA”) with certain accredited investors
(the “ANEW PIPE Investors”) for an aggregate purchase price of up to $2,000,000 (the “ANEW PIPE Financing”),
which included 900,000 bonus shares of common stock. Upon the closing of the ANEW PIPE Financing (funded and closed in connection
with the closing of the Merger on June 21, 2024), which totaled $1,950,000, of which $1,000,000 was used by the Company
to settle transaction costs. The Company received approximately $950,000 in cash proceeds.
Concurrent
with Closing, certain ANEW stockholders will be issued up to 5,000,000 additional shares of Redwoods’ Common Stock, now Public
ANEW, (the “Contingent Consideration Shares”), each valued at $10 per share, or an aggregate equity value of $50,000,000,
which will be issued as follows:
(i) 2,000,000
Contingent Consideration Shares upon Redwoods achieving a closing price equal to or exceeding $12.50 for 10 trading days within
a 20-day trading period in the first three years following the Closing;
(ii) 2,000,000
Contingent Consideration Shares upon Redwoods achieving a closing price equal to or exceeding $15.00 for 10 trading days within
a 20-day trading period in the first three years following the Closing; and
(iii) 1,000,000
Contingent Consideration Shares upon Redwoods achieving a closing price equal to or exceeding $20.00 for 10 trading days within
a 20-day trading period in the first five years following the Closing.
Assuming
all the conditions for the issuance of the Contingent Consideration Shares are satisfied, the sum of the Merger Consideration and the
Contingent Consideration will be $110,000,000, assuming a price of $10 per share.
In
accordance with guidance applicable to these circumstances, the equity structure has been restated in all comparable periods up to June
21, 2024 and reflected as such as of June 30, 2024, to reflect the number of shares of the Company’s common stock, $0.0001 par
value per share, issued to ANEW’s stockholders in connection with the merger. As such, the shares and corresponding capital amounts
and earnings per share related to ANEW’s common stock prior to the merger have been retroactively restated as shares reflecting
the exchange ratio established in the merger.
For
accounting purposes, the Merger was treated as the equivalent of the Company issuing shares for the net assets of Redwoods,
accompanied by a recapitalization. The net assets of Redwoods were stated at historical cost with no goodwill or other intangible
assets recorded. In connection with the Merger, in addition to the warrants, Public ANEW assumed $589,081 in cash and $568,111 in
income tax payable.
NOTE 2 — SUMMARY
OF SIGNIFICANT ACCOUNTING POLICIES
Basis
of Presentation and Principles of Consolidation
The
Company prepares its consolidated financial statements in accordance with accounting principles generally accepted in the United States
of America (“U.S. GAAP”) and pursuant to the rules and regulations of the SEC. The Company prepared the Financial Statements,
without audit, pursuant to the rules and regulations of the SEC applicable to quarterly reporting on Form 10-Q and reflect, in management’s
opinion, all adjustments necessary to present fairly the financial information. All such adjustments are of a normal recurring nature.
Certain information and footnote disclosures normally included in financial statements, prepared in accordance with generally accepted
accounting principles, have been consolidated or omitted as permitted by such rules and regulations. These Financial Statements should
be read in conjunction with the consolidated financial statements and related notes included in the 2023 Annual Report. Results of operations
for interim periods are not necessarily indicative of annual results.
Reclassification
Certain
prior year amounts have been reclassified for comparative purposes to conform to the current-year financial statement presentation. These
reclassifications had no effect on previously reported results of operations and were not material.
Emerging
Growth Company
The
Company is an “emerging growth company,” as defined in Section 2(a) of the Securities Act of 1933, as amended (the “Securities
Act”), as modified by the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), and it may take advantage of
certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies
including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley
Act of 2002, reduced disclosure obligations regarding executive compensation in its periodic reports and proxy statements, and exemptions
from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute
payments not previously approved. Further, Section 102(b)(1) of the JOBS Act exempts emerging growth companies from being required to
comply with new or revised financial accounting standards until private companies (that is, those that have not had a Securities Act
registration statement declared effective or do not have a class of securities registered under the Securities Exchange Act of 1934,
as amended (the “Exchange Act”)) are required to comply with the new or revised financial accounting standards. The JOBS
Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging
growth companies but any such election to opt out is irrevocable. The Company has elected not to opt out of such extended transition
period which means that when a standard is issued or revised and it has different application dates for public or private companies,
the Company, as an emerging growth company, can adopt the new or revised standard at the time private companies adopt the new or revised
standard. This may make comparison of the Company’s financial statements with another public company which is neither an emerging
growth company nor an emerging growth company which has opted out of using the extended transition period difficult or impossible because
of the potential differences in accounting standards used.
Use
of Estimates
The
preparation of unaudited consolidated financial statements in conformity with U.S. GAAP requires the Company’s management to make
estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities
at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period.
Making
estimates requires management to exercise significant judgment. It is at least reasonably possible that the estimate of the effect of
a condition, situation or set of circumstances that existed at the date of the financial statements, which management considered in formulating
its estimate, could change in the near term due to one or more future confirming events. Accordingly, the actual results could differ
significantly from those estimates.
Cash
and Cash Equivalents
Cash
and cash equivalents represent cash on hand, demand deposits, and other short-term highly liquid investments placed with banks,
which have original maturities of three months or less and are readily convertible to known amounts of cash.
Concentration
of Credit Risk
Financial
instruments that potentially subject the Company to concentrations of credit risk consist of a cash account in a financial institution,
which, at times, may exceed the Federal Depository Insurance Coverage of $250,000. As of June 30, 2024, the Company has not experienced
losses on this account and management believes the Company is not exposed to significant risks on such account.
Fair
Value of Financial Instruments
The
assets and liabilities are valued using a fair market basis as defined in the Financial Accounting Standards Board (“FASB”)
Accounting Standards Update (“ASU”) ASC 820, Fair Value Measurement. Fair value is the price the Company would receive
to sell an asset or pay to transfer a liability in an orderly transaction with a market participant at the measurement date. The Company
uses a three-level hierarchy established by the FASB that prioritizes fair value measurements based on the types of inputs used
for the various valuation techniques (market approach, income approach and cost approach). The levels of the fair value hierarchy are
described below:
| |
Level 1: |
Quoted prices in active
markets for identical assets or liabilities. |
| |
|
|
| |
Level 2: |
Inputs other than quoted
prices that are observable for the asset or liability, either directly or indirectly; these include quoted prices for similar assets
or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. |
| |
|
|
| |
Level 3: |
Unobservable inputs with
little or no market data available, which require the reporting entity to develop its own assumptions. |
The
Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment
and considers factors specific to the asset or liability. Financial assets and liabilities are classified in their entirety based on
the most conservative level of input that is significant to the fair value measurement.
| | |
| | |
Fair
value measurements at reporting date using: | |
| | |
Fair
value | | |
Quoted
prices
in active
markets for
identical
liabilities
(Level 1) | | |
Significant
other
observable
inputs
(Level 2) | | |
Significant
unobservable
inputs
(Level 3) | |
| Liabilities: | |
| | |
| | |
| | |
| |
| Public warrant liabilities, June 30, 2024 | |
$ | 488,750 | | |
$ | 488,750 | | |
$ | - | | |
$ | - | |
| Private warrant liabilities, June 30, 2024 | |
$ | 62,222 | | |
$ | - | | |
$ | - | | |
$ | 62,222 | |
| | |
| | | |
| | | |
| | | |
| | |
| Liabilities: | |
| | | |
| | | |
| | | |
| | |
| Public warrant liabilities, December 31, 2023 | |
$ | - | | |
$ | - | | |
$ | - | | |
$ | - | |
| Private warrant liabilities, December 31, 2023 | |
$ | - | | |
$ | - | | |
$ | - | | |
$ | - | |
The
following tables present a reconciliation of the Level 3 Private Warrants liabilities:
| | |
Six
months ended
June 30, | |
| | |
2024 | | |
2023 | |
| Private warrant liabilities, January 1 | |
$ | - | | |
$ | - | |
| Issuances/Assumptions | |
| 22,525 | | |
| - | |
| Exercises | |
| - | | |
| - | |
| Change in fair value | |
| 39,697 | | |
| - | |
| Private warrant liabilities, June 30 | |
$ | 62,222 | | |
$ | - | |
| | |
Three
months ended
June 30, | |
| | |
2024 | | |
2023 | |
| Private warrant liabilities, April 1 | |
$ | - | | |
$ | - | |
| Issuances/Assumptions | |
| 22,525 | | |
| - | |
| Exercises | |
| - | | |
| - | |
| Change in fair value | |
| 39,697 | | |
| - | |
| Private warrant liabilities, June 30 | |
$ | 62,222 | | |
$ | - | |
Intangible
Assets
The
Company’s intangible assets consist of acquired medical licenses and patents.
The
Company acquires medical licenses for the treatment of medical conditions to market and sell in the future. The initial asset cost is
the cost to acquire the license. Once in use, the Company amortizes the license cost over the useful life using the straight-line method.
The
Company records the cost to acquire patents as the initial asset cost. Once the patents are approved and in use, and assuming no litigations
expenses, the Company amortizes the patent cost over the useful life using the straight-line method. The amortization period will not
exceed the lifespan of the protection afforded by the patent. If the expected useful life of the patent is even shorter, the Company
will use the useful life for amortization purposes. Thus, the shorter length of a patent’s useful life and its legal life will
be used for the amortization period.
Impairment
of Long-Lived and Intangible Assets
The
Company assesses the impairment of long-lived and intangible assets periodically, or at least annually, and whenever events or changes
in circumstances indicate that the carrying value may not be recoverable. Factors considered important, which could trigger an impairment
review, include the following: significant underperformance relative to historical or projected future cash flows; significant changes
in the manner of use of the assets or the strategy of the overall business; and significant negative industry trends. When management
determines that the carrying value of long-lived and intangible assets may not be recoverable, impairment is measured as the excess of
the assets’ carrying value over the estimated fair value. Management is not aware of any other impairment changes that may currently
be required; however, the Company cannot predict the occurrence of events that might adversely affect the reported values in the future.
On an annual basis, the Company tests the long-lived and intangible assets for impairment based on the projected net present value of
cash flows for each asset. Prior to the annual impairment test, if circumstances change and a long-lived or intangible asset is deemed
impaired, an impairment loss will be immediately recognized in the statements of operations. At December 31, 2023, the date of the last
impairment test, it was determined that the estimated fair value of the intangible assets exceeded the carrying value of the assets by
50%, indicating no impairment.
Revenue
Recognition
The
Company is in a pre-revenue state and does not generate revenue. When the Company commences to derive revenue, those contracts will be
accounted in accordance with ASU 2014-09, Revenue from Contracts with Customers (Topic ASC 606).
Income
Taxes
The
Company uses the asset and liability method of accounting for income taxes in accordance with ASU 740, “Income Taxes”. Under
this method, income tax expense is recognized as the amount of: (i) taxes payable or refundable for the current year and (ii) future
tax consequences attributable to differences between the consolidated financial statements carrying amounts of existing assets and
liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply
to taxable income in the years which those temporary differences are expected to be recovered or settled. The effect on deferred
tax assets and liabilities of a change in tax rates is recognized in the results of operations in the period that includes the enactment
date. A valuation allowance is provided to reduce the deferred tax assets reported if based on the weight of available evidence it is
more likely than not that some portion or all of the deferred tax assets will not be realized.
The
Company is subject to Income tax filings requirements in U.S. federal and various state jurisdictions. The Company’s tax returns
for years from 2021, 2022, and 2023 are subject to U.S. federal, state, and local income tax examinations by tax authorities.
The
Company reports income tax related interest and penalties within the income tax line item on the consolidated statements of operations.
The Company likewise reports the reversal of income tax-related interest and penalties within such line item to the extent the Company
resolves the liabilities for uncertain tax positions in a manner favorable to the accruals.
Net
Loss Per Share (Basic and Diluted)
Basic
net loss per share is computed by dividing net loss by the weighted average number of shares outstanding during the period. Diluted net
loss per share is computed by dividing net loss by the weighted average number of shares outstanding, plus the number of additional shares
that would have been outstanding if the common share equivalents had been issued, if dilutive.
The
following table details the net loss per share calculation, reconciles between basic and diluted weighted average shares outstanding,
and presents the potentially dilutive shares that are excluded from the calculation of the weighted average diluted common shares outstanding,
because their inclusion would have been anti-dilutive:
| | |
For
the Six Months Ended
June 30, | |
| | |
2024 | | |
2023 | |
| Numerator: | |
| | |
| |
| Net
loss | |
$ | (1,123,683 | ) | |
$ | (436,872 | ) |
| | |
| | | |
| | |
| Weighted
average shares outstanding (denominator for basic earnings per share) | |
| 15,678,898 | | |
| 15,130,393 | |
| | |
| | | |
| | |
| Weighted
average shares and assumed potential common shares (denominator for diluted earnings per share, treasury method) | |
| 15,678,898 | | |
| 15,130,393 | |
| | |
| | | |
| | |
| Basic
loss per share | |
$ | (0.07 | ) | |
$ | (0.03 | ) |
| Diluted
loss per share | |
$ | (0.07 | ) | |
$ | (0.03 | ) |
The
following common share equivalents are excluded from the calculation of weighted average common shares outstanding, because their inclusion
would have been anti-dilutive:
| | |
For
the Six Months Ended
June 30, | |
| | |
2024 | | |
2023 | |
| Warrants | |
| 12,030,000 | | |
| 12,030,000 | |
| Total potentially
dilutive shares | |
| 12,030,000 | | |
| 12,030,000 | |
Research
and Development Cost
Research
and development (R&D) costs are expensed as incurred. R&D costs are related to the Company’s internally funded development
of the Company medical licenses and patents. The Company R&D costs were $0 for the three and six months ended June
30, 2024 and 2023, respectively.
Share-based
Compensation
The
Company accounts for share-based compensation in accordance with the fair value recognition provisions of the Financial Accounting Standards
Board (“FASB”) Accounting Standards Codification (“ASC”) No. 718 and No. 505. The Company issues restricted stock
to employees and consultants for their services. Cost for these transactions are measured at the fair value of the equity instruments
issued at the date of grant. These shares are considered fully vested and the fair market value is recognized as an expense in the period
granted. The Company recognized consulting expenses and a corresponding increase to additional paid-in-capital related to stock issued
for services. For agreements requiring future services, the consulting expense is to be recognized ratably over the requisite service
period.
The
Company recorded share-based compensation of $37,514 and $0 for the six months ended June 30, 2024, and 2023, respectively.
Related
Parties
The
Company follows subtopic 850-10 of the FASB Accounting Standards Codification for the identification of related parties and disclosure
of related party transactions.
Pursuant
to Section 850-10-20 the related parties include (a) affiliates of the Company; (b) entities for which investments in their equity securities
would be required, absent the election of the fair value option under the Fair Value Option Subsection of Section 825–10–15,
to be accounted for by the equity method by the investing entity; (c) trusts for the benefit of employees, such as pension and profit-sharing
trusts that are managed by or under the trusteeship of management; (d) principal owners of the Company; (e) management of the Company;
(f) other parties with which the Company may deal if one party controls or can significantly influence the management or operating policies
of the other to an extent that one of the transacting parties might be prevented from fully pursuing its own separate interests; and
(g) other parties that can significantly influence the management or operating policies of the transacting parties or that have an ownership
interest in one of the transacting parties and can significantly influence the other to an extent that one or more of the transacting
parties might be prevented from fully pursuing its own separate interests.
The
financial statements shall include disclosures of material related party transactions, other than compensation arrangements, expense
allowances, and other similar items in the ordinary course of business. However, disclosure of transactions that are eliminated in the
preparation of consolidated or combined financial statements is not required in those statements. The disclosures shall include: (a)
the nature of the relationship(s) involved; (b) description of the transactions, including transactions to which no amounts or nominal
amounts were ascribed, for each of the periods for which income statements are presented, and such other information deemed necessary
to an understanding of the effects of the transactions on the financial statements; (c) the dollar amounts of transactions for each of
the periods for which income statements are presented and the effects of any change in the method of establishing the terms from that
used in the preceding period; and (d) amounts due from or to related parties as of the date of each balance sheet presented and, if not
otherwise apparent, the terms and manner of settlement.
Recent
Accounting Pronouncements
Management
does not believe that any recently issued, but not yet effective, accounting pronouncements, if currently adopted, would have a material
effect on the Company’s consolidated financial statements.
NOTE
3 — GOING CONCERN
The
accompanying consolidated financial statements have been prepared as if the Company will continue as a going concern. The Company has
incurred significant operating losses and negative cash flows from operations since inception. As of June 30, 2024, the Company had cash
of approximately $845,000 and an accumulated deficit of approximately $5.5 million. The Company has incurred recurring losses,
has experienced recurring negative operating cash flows, and requires significant cash resources to execute its business plans. The Company
is dependent on obtaining additional working capital funding from the sale of equity and/or debt securities in order to continue to execute
its development plans and continue operations. Without additional funding, there is substantial doubt about the Company’s ability
to continue as a going concern for twelve months from the date of these financial statements.
NOTE 4 — PREPAID
EXPENSES
Prepaid
expenses consist of the D&O insurance. As of June 30, 2024 and December 31, 2023, the prepaid expenses, net were $154,500 and $0,
respectively, in the accompanying consolidated balance sheet.
NOTE 5 — INTANGIBLE
ASSETS
Licenses
During
2015, the Company acquired two licenses for two licensed platform technologies, a biosimilar biologics platform that uses biologic therapies
to treat cancer – recombinant antibodies, and a gene therapy platform which uses a gene therapy approach to introduce a therapeutic
protein called “Klotho” inside the body to treat neurodegenerative diseases. The value of the licenses was $736,983 at June
30, 2024.
On
September 12, 2022, the Company acquired four market-approved anti-cancer drugs approved for sale in Germany for $1,308,270.
The purchase price represents the fair value of the intangible asset based on the net present value of the projected gross profit to
be generated by the licenses. The value of the licenses was $1,308,270 at June 30, 2024.
On
January 24, 2022, the Company signed an exclusive, world-wide License Agreement with the University of Barcelona for a cell and/or gene
therapy that has shown compelling activity in animal models of human Alzheimer’s disease and amyotrophic lateral sclerosis (“ALS”
or “Lou Gehrig’s disease”). The gene therapy will also be applied to age-related diseases and rare (“Orphan”)
diseases. Beginning on December 15, 2022, the Quarterly license fee is 10,000 Euros. In addition, the Company will pay a Royalty equal
to 3% of net sales of finished products. For the six months ended June 30, 2024 and 2023, the Company owes $0 under the agreement.
On
January 27, 2023, the Company signed a License Agreement with Teleost Biopharmaceutic, LLC to acquire various assets for the Company’s
proprietary pharmaceutical program segment. The license includes the use of patented small drug molecules that bind to the melanocortin
receptors on human cells and affect skin pigmentation. The terms include a $10,000 fee for signing the agreement and a $50,000 payment
on January 27, 2024. The Company will pay for all new patent costs for new discoveries and new treatments. The Company will make standard
commercial development-based milestone payments for the various stages of license development and regulatory approval. In addition, the
Company will make royalty payments on the net sales for commercial products. Beginning in 2025, the Company will also pay patent and
license maintenance fees. The amount due under the agreement was $10,000 at June 30, 2024.
On
March 5, 2023, the Company signed a Non-Exclusive License Agreement with Heidelberg University to grant non-exclusive rights to various
licenses owned and under development by the university. The licenses include the use of modified AAV capsid polypeptides for treatment
of muscular diseases. The terms include a €50,000 ($56,325) fee for signing the agreement and €100,000 ($112,650) payment within
60 days of the anniversary of signing the agreement. The Company will pay €1,000,000 ($1,126,500) for each assignment of a right
to a license owned by the university. For new licenses, the Company will make standard commercial development-based milestone payments
for the various stages of license development and regulatory approval. The Company will make 2 % royalty payments by January 31st each
year during the term of the agreement for each licensed product for the proceeding calendar year. At June 30, 2024, the Company paid
$179,821 under the agreement.
On
December 1, 2023, the Company signed a license agreement with TransferTech Sherbooke for the rights to develop and commercialize the
technology of a “Needleless Syringe.” Under the terms of the agreement, the Company paid a $26,060 upfront fee and royalty
fees on the license income. The Company has not commenced developing the technology. The amount paid was $26,060 at June 30, 2024.
The
total licenses recorded were $2,261,134 and $2,137,638 at June 30, 2024 and December 31, 2023, respectively, in the accompanying consolidated
balance sheet. The licenses are not in use. Once the licenses are in use, the licenses will be amortized over the useful life.
Patents
The Company acquires patents for Alzheimer, ALS and other items from
third parties. Once the patents are declared effective, patents are amortized using the straight-line method over their estimated
useful lives or statutory lives, whichever is shorter, and will be reviewed for impairment upon any triggering event that may impact the
assets’ ultimate recoverability as prescribed under the guidance related to impairment of long-lived assets. Costs incurred to acquire patents,
including legal costs, are also capitalized as long-lived assets and amortized on a straight-line basis with the associated patent. At
December 31, 2023, certain professional fees incurred for the patents in the amount of $47,740 were deemed not capitalizable and were
expensed as professional fees in the accompanying statements for operations. At June 30, 2024, professional fees incurred for the patents
in the amount of $30,898 were deemed not capitalizable and were expensed as professional fees in the accompanying statements for operations.
The patent value as of June 30, 2024 and December 31, 2023 was $48,420, respectively, in the accompanying consolidated balance sheet.
NOTE
6 — ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts
payable and accrued expenses consist of professional fees. The accounts payable and accrued expenses as of June 30, 2024 and December
31, 2023 were $284,388 and $153,719, respectively, in the accompanying consolidated balance sheet.
NOTE 7
— COMMITMENTS AND CONTINGENCIES
From
time to time, the Company is subject to various legal proceedings and claims, either asserted or unasserted, that arise in the ordinary
course of business. Although the outcome of the various legal proceedings and claims cannot be predicted with certainty, management does
not believe that any of these proceedings or other claims will have a material effect on the Company’s business, financial condition,
results of operations or cash flows.
Material
Contracts
On
November 27, 2014, the Company signed a License Agreement and a Manufacturing and Supply Agreement for the monoclonal antibody development
license and supply agreement and related manufacturing with Reliance Life Sciences (RLS), the life science arm of Reliance Industries
Pvt Ltd, the largest private company in India. The contract expires on November 27, 2024 with a 10-year renewal option. The License Agreement
entitles the Company to pay $100,000 per product for a total of three products with milestone payments for meeting certain criteria.
In addition, the Company will pay a quarterly royalty payment of 5% on net sales of finished products. The Manufacturing and Supply Agreement
contains an estimated acquisition price of active pharmaceutical ingredients (API) of $350,000 per Kg for each product developed.
As of June 30, 2024, the Company has not generated any activity under the agreement.
On
October 19, 2022, the Company signed an M&A/Capital Markets Advisory Agreement with Chardan Capital Markets to advise and assist
the Company in negotiating the terms and conditions with respect to a potential sale, purchase, merger, joint venture, business combination,
material change of control, or similar transaction involving the Company and a strategic acquirer and/or private or publicly listed entity
or business, including a Special Purpose Acquisition Company (SPAC), and with respect to any offerings of any equity, equity-linked or
debt securities of the Company or any other party to a financing transaction and perform such other financial advisory services to the
Company. At the close of the merger on June 21, 2024, the Company paid $3.0 million and 1.5 million in common shares for M&A advisory
fees and deferred underwriting fees.
On
June 13, 2024, RWOD and ANEW entered into a forward purchase agreement with (i) Meteora Capital Partners, LP (“MCP”),
(ii) Meteora Select Trading Opportunities Master, LP (“MSTO”), and (iii) Meteora Strategic Capital, LLC
(“MSC” and, collectively with MCP and MSTO, the “Seller”) (the “Forward Purchase Agreement”). Redwoods
is the holder of the asset and Sponsor and is also a counterparty to Public ANEW. Upon Closing of the merger on June 21, 2024 and on
June 30, 2024, the value of the contract for the Company was $0 as the contract created no receivable or obligation for the Company.
The Company will assess the Company obligation and value the contract in the future periods based on fair value and record changes on
the fair value in the Consolidated Statements of Operations.
NOTE 8 — NOTES
PAYABLE
On
September 12, 2022, the Company issued a $1,308,270 promissory note used to acquire four market-approved anti-cancer drugs. See Note
5 – Intangible Assets for further discussion. The promissory note bore interest at 6% and had a maturity date of June
30, 2023. By agreement, the interest stopped accruing at June 30, 2023. As of December 31, 2023, the Company made interest payments of
$78,496 to fully satisfy the interest obligation under the promissory note. The note was paid off as part of the merger that closed on
June 21, 2024. The unpaid principal balance of the note was $0 and $1,308,270 at June 30, 2024 and December 31, 2023, respectively.
On March 4, 2024, in
connection with the Merger, Public ANEW entered into a convertible promissory note and Securities Purchase Agreement (“SPA”)
with certain accredited investors (the “Redwoods PIPE Investors”) for an aggregate purchase price of up to $2,000,000
(the “Redwoods PIPE Financing”), which included 750,000 bonus shares of common stock. Upon the closing of the Redwoods
PIPE Financing (funded and closed in connection with the closing of the Merger on June 21, 2024), which totaled $1,950,000,
of which $1,768,661 was used by the Company to settle transaction costs. The Company received approximately $181,339 in net cash proceeds.
On April 22, 2024, in connection with the Merger,
Public ANEW entered convertible promissory note and Securities Purchase Agreement (“SPA”) with certain accredited investors
(the “ANEW PIPE Investors”) for an aggregate purchase price of up to $2,000,000 (the “ANEW PIPE Financing”),
which included 900,000 bonus shares of common stock. Upon the closing of the ANEW PIPE Financing (funded and closed in connection
with the closing of the Merger on June 21, 2024), which totaled $1,950,000, of which $1,000,000 was used by the Company
to settle transaction costs. The Company received approximately $950,000 in cash proceeds.
Both
convertible promissory notes, Redwoods PIPE Financing and ANEW PIPE Financing bare an interest rate of 10% as of June 30, 2024. The total
accrued interest for both convertible promissory notes at June 30, 2024 is approximately $15,064.
NOTE 9 — RELATED
PARTIES
On
October 10, 2021, the Company signed an Employment Agreement with Dr, Joseph Sinkule to serve as the Company’s CEO for three years
ending on October 9th, 2024. In addition, Mr. Sinkule will serve as a member of the board of directors for a five-year term.
Mr. Sinkule’s annual salary will be $240,000 per year and increase to $360,000 per year upon raising a total of five million dollars
($5,000,000) or more in equity and/or debt financing. The Company’s CEO has earned $240,000 for the years ended December 31, 2023
and 2022. In accordance with the agreement, at June 30, 2024 and December 31, 2023, the Company’s CEO is owed $0 and $80,000, respectively.
During
November 2022, the Company advanced a shareholder $300,000 as a short-term loan. The loan is non-interest bearing and due by the end
of December 2022. The shareholder repaid $50,000 during December 2022 and $250,000 in January 2023 to fully satisfy the advance. At June
30, 2024 and December 31, 2023, the loan balance was $0, respectively.
On
December 12, 2023, the Company issued a promissory note to a member of management. The promissory note accrued interest at a one-time
interest fee of $2,460, which was paid off as of June 30, 2024. The unpaid principal balance was $31,000 and $24,000 at June 30, 2024
and December 31, 2023, respectively.
At
June 30, 2024 and December 31, 2023, the aggregate related party payable was $31,000 and $135,000, respectively.
NOTE 10 —
STOCKHOLDER’S EQUITY (DEFICIT)
On
June 21, 2024, the Business Combination, among other transactions contemplated by the Merger Agreement, was completed. The transaction
was accounted as a reverse recapitalization in accordance with GAAP. Under this method of accounting, Redwoods was treated as the
“acquired” company for financial reporting purposes. Accordingly, for accounting purposes, the financial statements of the
Combined Company represent a continuation of the financial statements of ANEW with the Transactions treated as the equivalent of ANEW
issuing shares for the net assets of Redwoods, accompanied by a recapitalization. Under this method of accounting, Redwoods
was treated as the acquired company for financial reporting purposes. Accordingly, for accounting purposes, the Merger was
treated as the equivalent of the Company issuing shares for the net assets of Redwoods, accompanied by a recapitalization.
The net assets of Redwoods were stated at historical cost with no goodwill or other intangible assets recorded. See “NOTE 1 — Organization
and Business Description” for detail.
Equity
Incentive Compensation
In
connection with the Business Combination, the Public ANEW Board adopted, and the Company’s stockholders approved, the 2024 Equity
Incentive Plan (“Equity Incentive Plan”). Although Public ANEW does not have a formal policy with respect to the grant of
equity incentive awards to Public ANEW’s executive officers, the Company believes that equity awards provide Public ANEW’s
executive officers with a strong link to Public ANEW’s long-term performance, create an ownership culture and help to align the
interests of Public ANEW’s executives and Public ANEW’s stockholders. In addition, Public ANEW believes that equity awards
with a time-based vesting feature promote executive retention because this feature provides incentives to Public ANEW’s executive
officers to remain in Public ANEW’s employment during the applicable vesting period. Accordingly, Public ANEW’s board of
directors periodically reviews the equity incentive compensation of Public ANEW’s executive officers and from time to time may
grant equity incentive awards to them. No stock options or other equity awards were granted to Public ANEW executive officers during
the fiscal year ended December 31, 2023 and as of June 30, 2024.
NOTE 11 — SUBSEQUENT
EVENTS
The
Company has evaluated subsequent events pursuant to the requirements of ASC Topic 855, from the balance sheet date through the date the
financial statements were issued, and has determined that the following subsequent event exists:
On
August 12, 2024, ANEW PIPE Investor converted $2,000,000 of the principal amount and related interest of the ANEW PIPE Financing note
issued on April 22, 2024 funded in connection with the Merger on June 21, 2024 into 1,550,617 shares of the Company’s
common stock, with remaining principal balance due of $0.

REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Strategic Asset Leasing,
Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated
balance sheets of Strategic Asset Leasing, Inc. (the “Company”) as of December 31, 2023, and 2022, the related statements of
operations, stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as
the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial
position of the Company as of December 31, 2023, and 2022, and the results of its operations and its cash flows for the years then ended,
in conformity with accounting principles generally accepted in the United States.
Going Concern Considerations
The accompanying financial statements have been
prepared assuming that the Company will continue as a going concern. The Company has suffered recurring losses since inception and has
not achieved profitable operations, which raises substantial doubt about its ability to continue as a going concern. Management’s
plans in regard to these matters are described in Note 2. The financial statements do not include any adjustments that might result from
the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility
of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our
audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are
required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and
regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the
standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial
statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged
to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding
of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s
internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess
the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond
to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements.
Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating
the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
There were no critical audit matters to be communicated
from the current period audit of the financial statements.

We have served as the Company’s auditor
since 2024.
Short Hills, New Jersey
May 22, 2024
PCAOB registration # 3313
STRATEGIC ASSET LEASING, INC.
CONSOLIDATED BALANCE SHEETS
| | |
December 31, 2023 | | |
December 31, 2022 | |
| | |
| | |
| |
| ASSETS | |
| | |
| |
| Current assets: | |
| | |
| |
| Cash | |
$ | 2,808 | | |
$ | 75,872 | |
| Prepaid expenses | |
| 3,840 | | |
| 3,667 | |
| Due from related party | |
| - | | |
| 250,000 | |
| Total current assets | |
| 6,648 | | |
| 329,539 | |
| | |
| | | |
| | |
| Other assets | |
| | | |
| | |
| Licenses | |
| 2,137,638 | | |
| 2,123,750 | |
| Patents | |
| 48,420 | | |
| 86,160 | |
| Total other assets | |
| 2,186,058 | | |
| 2,209,910 | |
| | |
| | | |
| | |
| Total Assets | |
$ | 2,192,706 | | |
$ | 2,539,449 | |
| | |
| | | |
| | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | |
| | | |
| | |
| | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable | |
$ | 151,259 | | |
$ | 8,014 | |
| Accrued expenses | |
| 2,460 | | |
| 4,742 | |
| Related party payable | |
| 135,000 | | |
| - | |
| Note payable | |
| 1,332,270 | | |
| 1,347,518 | |
| Total current liabilities | |
| 1,620,989 | | |
| 1,360,274 | |
| | |
| | | |
| | |
| Commitments and contingencies | |
| - | | |
| - | |
| | |
| | | |
| | |
| Stockholders’ equity (deficiency): | |
| | | |
| | |
| Preferred stock Series B, $0.001 par value; 500,000
shares authorized;405,250 issued and outstanding as of December 31, 2023 and December 31, 2022 | |
| 405 | | |
| 405 | |
| Preferred stock Series C, $0.0001 par value; 5,000,000
shares authorized; 1,000,000 issued and outstanding as of December 31, 2023 and December 31, 2022 | |
| 100 | | |
| 100 | |
| Common stock, $0.0001 par value; 1,500,000,000 shares
authorized; 1,044,861,360 issued and outstanding as of December 31, 2023 and December 31, 2022 | |
| 104,486 | | |
| 104,486 | |
| Additional paid in capital | |
| 3,539,003 | | |
| 3,539,003 | |
| Common stock to be issued | |
| 851,400 | | |
| 751,400 | |
| Accumulated deficit | |
| (3,923,677 | ) | |
| (3,216,219 | ) |
| Total stockholders’ equity (deficiency) | |
| 571,717 | | |
| 1,179,175 | |
| | |
| | | |
| | |
| Total Liabilities and Stockholders’ equity | |
$ | 2,192,706 | | |
$ | 2,539,449 | |
The accompanying notes are an integral part of these financial statements.
STRATEGIC ASSET LEASING, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| | |
For the Twelve Months Ended | |
| | |
December 31, 2023 | | |
December 31, 2022 | |
| | |
| | |
| |
| Operating expenses: | |
| | |
| |
| Professional fees | |
$ | 600,776 | | |
$ | 526,007 | |
| General and administrative | |
| 30,546 | | |
| 48,367 | |
| Total operating expenses | |
| 631,322 | | |
| 574,374 | |
| | |
| | | |
| | |
| Net operating income (loss) | |
| (631,322 | ) | |
| (574,374 | ) |
| | |
| | | |
| | |
| Other (income) expense: | |
| | | |
| | |
| Interest expense | |
| 76,214 | | |
| 24,366 | |
| Other (income) expense | |
| (78 | ) | |
| (147 | ) |
| Total Other (income) expense | |
| 76,136 | | |
| 24,219 | |
| | |
| | | |
| | |
| Net income (loss) | |
$ | (707,458 | ) | |
$ | (598,593 | ) |
| | |
| | | |
| | |
| Basic and diluted income (loss) per share | |
$ | (0.00 | ) | |
$ | (0.00 | ) |
| | |
| | | |
| | |
| Weighted average number of common shares outstanding -
basic | |
| 1,044,861,360 | | |
| 1,044,861,360 | |
The accompanying notes are an integral part of these financial statements.
STRATEGIC ASSET LEASING, INC.
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY
(DEFICIT)
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
Accumulated | | |
| |
| | |
Pref.
Stock - | | |
Pref.
Stock - | | |
| | |
| | |
Additional | | |
| | |
| | |
Deficit
During | | |
Total
Stockholders’ | |
| | |
Series
B | | |
Series
C | | |
Common
Stock | | |
Paid-In | | |
Stock to be | | |
Accumulated | | |
Development | | |
Equity | |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Issued | | |
Deficit | | |
Stage | | |
(Deficit) | |
| Balance
at December 31, 2021 | |
| 405,250 | | |
$ | 405 | | |
| 1,000,000 | | |
$ | 100 | | |
| 1,044,861,360 | | |
$ | 104,486 | | |
$ | 3,539,003 | | |
$ | - | | |
$ | (2,617,626 | ) | |
$ | - | | |
$ | 1,026,368 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock
subscription/compensation - common stock | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 751,400 | | |
| - | | |
| | | |
| 751,400 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (598,593 | ) | |
| - | | |
| (598,593 | ) |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Balance
at December 31, 2022 | |
| 405,250 | | |
$ | 405 | | |
| 1,000,000 | | |
$ | 100 | | |
| 1,044,861,360 | | |
$ | 104,486 | | |
$ | 3,539,003 | | |
$ | 751,400 | | |
$ | (3,216,219 | ) | |
$ | - | | |
$ | 1,179,175 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock
subscription & license purchase | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 100,000 | | |
| - | | |
| | | |
| 100,000 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (707,458 | ) | |
| - | | |
| (707,458 | ) |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Balance
at December 31, 2023 | |
| 405,250 | | |
$ | 405 | | |
| 1,000,000 | | |
$ | 100 | | |
| 1,044,861,360 | | |
$ | 104,486 | | |
$ | 3,539,003 | | |
$ | 851,400 | | |
$ | (3,923,677 | ) | |
$ | - | | |
$ | 571,717 | |
The accompanying notes are an integral part of these financial statements.
STRATEGIC ASSET LEASING, INC.
CONSOLIDATED STATEMENTS OF CASH FLOW
| | |
For the Twelve Months Ended | |
| | |
December 31, 2023 | | |
December 31, 2022 | |
| | |
| | |
| |
| Cash flows from operating activities: | |
| | | |
| | |
| Net income (loss) | |
$ | (707,458 | ) | |
$ | (598,593 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | | |
| | |
| Stock based compensation | |
| - | | |
| 1,400 | |
| Changes in operating assets and liabilities: | |
| | | |
| | |
| Prepaid expenses | |
| (173 | ) | |
| (7,122 | ) |
| Accounts payable | |
| 103,997 | | |
| 4,742 | |
| Accrued expenses | |
| (2,282 | ) | |
| - | |
| Related party payable | |
| 135,000 | | |
| - | |
| Net cash used in operating activities | |
| (470,916 | ) | |
| (599,573 | ) |
| | |
| | | |
| | |
| Cash flows from investing activities: | |
| | | |
| | |
| Patent acquisition costs (See Note 4) | |
| 37,740 | | |
| (86,160 | ) |
| Acquisition of licenses (See Note 4) | |
| (13,888 | ) | |
| (39,248 | ) |
| Net cash used in investing activities | |
| 23,852 | | |
| (125,408 | ) |
| | |
| | | |
| | |
| Cash flows from financing activities | |
| | | |
| | |
| Proceeds from stock subscriptions | |
| 100,000 | | |
| 750,000 | |
| Advance/Repayment of advance to shareholder | |
| 250,000 | | |
| (250,000 | ) |
| Proceeds from notes payable - current portion | |
| 24,000 | | |
| - | |
| Net cash provided by financing activities | |
| 374,000 | | |
| 500,000 | |
| | |
| | | |
| | |
| Net increase (decrease) in cash | |
| (73,064 | ) | |
| (224,981 | ) |
| Cash - beginning of the year | |
| 75,872 | | |
| 300,853 | |
| Cash - end of the year | |
$ | 2,808 | | |
$ | 75,872 | |
| | |
| | | |
| | |
| Supplemental disclosures: | |
| | | |
| | |
| Interest paid | |
$ | 78,496 | | |
$ | 19,624 | |
| Income taxes | |
$ | - | | |
$ | - | |
| | |
| | | |
| | |
| Supplemental disclosure for non-cash financing activities: | |
| | | |
| | |
| Acquisition of drugs licenses with a promissory note (See Note 5) | |
$ | - | | |
$ | 1,347,518 | |
The accompanying notes are an integral part of these financial statements.
Strategic Asset Leasing, Inc.
Notes to Consolidated Financial Statements
December 31, 2023
NOTE 1 – ORGANIZATION AND BASIS OF PRESENTATION
Organization
The accompanying consolidated financial statements
include Strategic Asset Leasing, Inc., formerly known as Mammoth Energy Group, Inc. (‘LEAS’ or the ‘Company’), its wholly owned subsidiary
and any majority controlling interests.
The Company was incorporated on February 27, 2006,
under the laws of the State of Nevada with the aim of pursuing lithium mining. Prior to being domiciled in Nevada, the Company was a Canadian
corporation known as Technigen Corporation. In March of 2013, management decided to change the domicile of the Company to Wyoming by filing
articles of continuance on March 5, 2013, subsequently dissolving the Nevada corporation.
On December 14, 2020, the Company entered a Stock
Purchase Agreement with Dr. Joseph Sinkule for 1,000,000 shares of the Company’s Series C preferred stock. The purchase price
was $110,000. Jason Tucker, the Company’s CEO, resigned from the Company and Mr. Simkule became the Company’s CEO and sole
director.
On November 1, 2021, the Company executed an Agreement
and Plan of Merger with Anew Acquisition Corp (“ANEW”), including the wholly own subsidiary ANEW Oncology, Inc., whereby each
issued and outstanding share of ANEW common stock was converted into the right to receive one-one hundredth (1/100) of a share of the
Company’s Series B preferred stock, par value $.001 per share.
After November 1, 2021, the Company will pursue
the development of its licensed rights in major world markets to biologic medicines and gene therapies that will be developed and commercialized
by the Company and affiliates and/or corporate partners.
On November 1, 2021, the shareholders of the Company
approved a name change to ANEW Medical, Inc. and approved a 1-for-2500 reverse split.
On January 4, 2022, the Company filed an Articles
of Amendment with the State of Wyoming, changing its name to “ANEW Medical, Inc.” and the contemplated 1-for-2,500 reverse
split. During January 2022 and in accordance with SEC Rule 10b-17 and FINRA Rule 6490, the Company submitted documents and other information
to FINRA in furtherance of pursuing and obtaining approval of the subject reverse stock split and name change. The Company must submit
the additional documents requested by, and necessary to obtain approval of, FINRA in connection with the subject reverse stock split and
name change. As of December 31, 2023, the reverse split and name change have not been declared effective by FINRA to broker deals in the
quotation system.
On May 30, 2023, the Company entered into a Business
Combination Agreement with Redwoods Acquisition Corp., a Delaware corporation (“Redwoods”), and Redwoods wholly owned subsidiary
ANEW Medical Sub, Inc., a Wyoming corporation (“Merger Sub”), The Business Combination Agreement and the transactions contemplated
thereby were approved by the board of directors of each of Redwoods and the Company.
The Business Combination Agreement provides, among
other things, on the Closing Date, upon the terms and conditions set forth herein and in accordance with the applicable provisions of
the Wyoming Business Corporations Act (the “WBCA”), Merger Sub will merge with and into the Company, with the Company
as the surviving company in the Merger and, after giving effect to such merger, a wholly owned Subsidiary of Redwoods, and each Company
Share will be converted into the right to receive the Merger Consideration, on the terms and subject to the conditions set forth in the
Business Combination Agreement.
The Business Combination is expected to close,
and the related S4 is expected to be effective, in April 2024, following the receipt of the required approval by the stockholders of Redwoods
and the Company, approval by the Nasdaq Stock Market (“Nasdaq”) of the initial listing application of the combined company
filed in connection with the Business Combination, and the fulfillment of other customary closing conditions.
Business
The Company was formed to develop essential
medicines for the treatment of chronic diseases – cancer, cardiovascular, and neurodegenerative disorders. The Company currently
has acquired two licensed platforms a generic drug portfolio and a biosimilar biologics platform that uses biologic therapies to treat
cancer, and two proprietary, patented technologies involving the melanocortin receptor-binding molecules and a gene therapy platform which
uses a gene therapy approach to introduce a therapeutic protein called “Klotho” inside the body to treat neurodegenerative
diseases.
On September 12, 2022, the Company acquired five
market-approved anti-cancer drugs approved for sale in Germany for $1,386,766. The Market Authorizations (MA’s) are for four of the
drugs that comprise the “FOLFOX” and “FOLFIRI” multi-drug regimens used in treatment of metastatic colorectal and
gastric cancer and in two of the drugs are used to treat metastatic lung cancer. The drugs are important in the treatment of many solid
tumors in both childhood and adult cancers. Previously, the Company acquired two off-patent biogeneric antibodies from Reliance Life Sciences
(RLS), the life science arm of Reliance Industries Pvt Ltd. of Navi Mumbai, India.
During January 2023, the Company acquired a treatment
for small drug molecules that bind to the melanocortin receptors on human cells and affect skin pigmentation for $20,000.
In accordance with Accounting Standards Codification
(“ASC”) 915, Development Stage Entities, the Company is considered to be in the development stage, with limited operations
since incorporating in the United States.
Summary of Significant Accounting Policies
Basis of Presentation
The financial statements of the Company have been
prepared in accordance with generally accepted accounting principles in the United States of America.
Use of Estimates
In preparing financial statements, management
makes estimates and assumptions that affect the reported amounts of assets and liabilities in the balance sheet and revenue and expenses
in the statement of expenses. Actual results could differ from those estimates.
Reclassifications
Certain prior year amounts have been reclassified
for comparative purposes to conform to the current-year financial statement presentation. These reclassifications had no effect on previously
reported results of operations. In addition, certain prior year amounts from the restated amounts have been reclassified for consistency
with the current period presentation.
Cash and Cash Equivalents
For the purposes of the statement of cash flows,
the Company considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents.
Concentrations of Risk
Cash and cash equivalents deposited with financial
institutions are insured by the Federal Deposit Insurance Corporation (“FDIC”). The Company did not hold cash in excess
of FDIC insurance coverage at a financial institution as of December 31, 2023 and 2022.
Prepaid Expenses
The Company considers all items incurred for future
services to be prepaid expenses. The prepaid expenses were $3,840 and $3,667 at December 31, 2023 and 2022, consisting of the OTC Market
annual fee.
Property and equipment
Property and equipment are recorded at cost and
depreciated on the straight-line method over the estimated useful lives. Expenditures for normal repairs and maintenance are charged to
expense as incurred. The cost and related accumulated depreciation of assets sold or otherwise disposed of are removed from the accounts,
and any gain or loss is included in operations.
Licenses
The Company acquires medical licenses for the
treatment of medical conditions to market and sell in the future. The initial asset cost is the cost to acquire the license. Once in use,
the Company amortizes the license cost over the useful life using the straight-line method.
Patents
The Company records the cost to acquire a commercial
license to technologies and patents as the initial asset cost. Once the patents are approved and
in use, and assuming no litigations expenses, the Company amortizes the patent cost over the useful life using the straight-line method.
The amortization period will not exceed the lifespan of the protection afforded by the patent. If the expected useful life of the patent
is even shorter, the Company will use the useful life for amortization purposes. Thus, the shorter length of a patent’s useful life and
its legal life will be used for the amortization period.
Valuation of Long-Lived and Intangible Assets
We assess the impairment of long-lived and intangible
assets periodically, or at least annually, and whenever events or changes in circumstances indicate that the carrying value may not be
recoverable. Factors considered important, which could trigger an impairment review, include the following: significant underperformance
relative to historical or projected future cash flows; significant changes in the manner of use of the assets or the strategy of the overall
business; and significant negative industry trends. When management determines that the carrying value of long-lived and intangible assets
may not be recoverable, impairment is measured as the excess of the assets’ carrying value over the estimated fair value. Management
is not aware of any other impairment changes that may currently be required; however, we cannot predict the occurrence of events that
might adversely affect the reported values in the future. On an annual basis, the Company tests the long-lived and intangible assets for
impairment based on the projected net present value of cash flows for each asset. Prior to the annual impairment test, if circumstances
change and a long-lived or intangible asset is deemed impaired, an impairment loss will be immediately recognized in the statements of
operations. At December 31, 2022, the date of the last impairment test, it was determined the estimated fair value exceeded the carry
value by in excess of 50%.
Derivative Financial Instruments
The Company does not use derivative instruments
to hedge exposures to cash flow, market, or foreign currency risks. The Company evaluates all of its financial instruments to determine
if such instruments are derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that
are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting
date, with changes in the fair value reported as charges or credits to income. For option-based derivative financial instruments, The
Company uses the Black-Scholes option-pricing model to value the derivative instruments at inception and subsequent valuation dates. The
classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is reassessed
at the end of each reporting period. Derivative instrument liabilities are classified in the balance sheet as current or non-current based
on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date.
Fair Value Measurements
In September 2006, the FASB issued ASC 820 (previously
SFAS 157) which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
The provisions of ASC 820 were effective January 1, 2008.
As defined in ASC 820, fair value is the price
that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement
date (exit price). The Company utilizes market data or assumptions that market participants would use in pricing the asset or liability,
including assumptions about risk and the risks inherent in the inputs to the valuation technique. These inputs can be readily observable,
market corroborated, or generally unobservable. The Company classifies fair value balances based on the observations of those inputs.
ASC 820 establishes a fair value hierarchy that prioritizes the inputs used to measure fair value. The hierarchy gives the highest priority
to unadjusted quoted prices in active markets for identical assets or liabilities (level 1 measurement) and the lowest priority to unobservable
inputs (level 3 measurement).
The three levels of the fair value hierarchy defined
by ASC 820 are as follows:
Level 1 – Quoted prices are available in
active markets for identical assets or liabilities as of the reporting date. Active markets are those in which transactions for the asset
or liability occur in sufficient frequency and volume to provide pricing information on an ongoing basis. Level 1 primarily consists of
financial instruments such as exchange-traded derivatives, marketable securities and listed equities.
Level 2 – Pricing inputs are other than
quoted prices in active markets included in level 1, which are either directly or indirectly observable as of the reported date. Level
2 includes those financial instruments that are valued using models or other valuation methodologies. These models are primarily industry-standard
models that consider various assumptions, including quoted forward prices for commodities, time value, volatility factors, and current
market and contractual prices for the underlying instruments, as well as other relevant economic measures. Substantially all of these
assumptions are observable in the marketplace throughout the full term of the instrument, can be derived from observable data or are supported
by observable levels at which transactions are executed in the marketplace. Instruments in this category generally include non-exchange-traded
derivatives such as commodity swaps, interest rate swaps, options and collars.
Level 3 – Pricing inputs include significant
inputs that are generally less observable from objective sources. These inputs may be used with internally developed methodologies that
result in management’s best estimate of fair value.
The Company did not identify any assets or liabilities
that are required to be adjusted on the balance sheet to fair value as of December 31, 2023 and 2022.
Revenue Recognition
Revenue is recognized when a customer obtains
control of promised goods or services and is recognized in an amount that reflects the consideration that an entity expects to receive
in exchange for those goods or services. In addition, the standard requires disclosure of the nature, amount, timing, and uncertainty
of revenue and cash flows arising from contracts with customers. The amount of revenue that is recorded reflects the consideration that
the Company expects to receive in exchange for those goods. The Company applies the following five-step model in order to determine this
amount: (i) identification of the promised goods in the contract; (ii) determination of whether the promised goods are performance
obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including
the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition
of revenue when (or as) the Company satisfies each performance obligation.
The Company only applies the five-step model to contracts
when it is probable that the entity will collect the consideration it is entitled to in exchange for the goods and service transfers to
the customer. Once a contract is determined to be within the scope of ASC 606 at contract inception, the Company reviews the contract
to determine which performance obligations the Company must deliver and which of these performance obligations are distinct. The Company
recognizes as revenues the amount of the transaction price that is allocated to the respective performance obligation when the performance
obligation is satisfied or as it is satisfied. Generally, the Company’s performance obligations are transferred to customers at
a point in time, typically upon delivery.
Income taxes
The Company’s policy is to provide for deferred
income taxes based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates that
will be in effect when the differences are expected to reverse. The U.S. Tax Cuts and Jobs Act (TCJA) legislation reduces the U.S. federal
corporate income tax rate from 35.0% to 21.0% and is effective June 22, 2018, for the Company. On January 1, 2023, the U.S. federal corporate
income tax increased from 21% to 28%. We did not provide any current or deferred U.S. federal income tax provision or benefit for any
of the periods presented because we have experienced operating losses since inception. When it is more likely than not that a tax asset
cannot be realized through future income the Company must allow for this future tax benefit. We provided a full valuation allowance on
the net deferred tax asset, consisting of net operating loss carryforwards, because management has determined that it is more likely than
not that we will not earn income sufficient to realize the deferred tax assets during the carryforward period.
The Company is not aware of any uncertain tax
position that, if challenged, would have a material effect on the financial statements for the three-months ended December 31, 2023, or
during the prior three years applicable under FASB ASC 740. We did not recognize any adjustment to the liability for uncertain tax
position and therefore did not record any adjustment to the beginning balance of accumulated deficit on the consolidated balance sheet.
The Company is in the process of filing all unfiled tax returns. All tax returns for the Company remain open for examination.
Basic and diluted net income per share
Basic net loss per common share is computed using
the weighted average number of common shares outstanding. Diluted earnings per share (EPS) include additional dilution from common stock
equivalents, such as stock issuable pursuant to convertible notes. Common stock equivalents are not included in the computation of diluted
earnings per share when the Company reports a loss because to do so would be anti-dilutive for the periods presented. On December 31,
2023 the Company’s common stock equivalents consisted of 405,250 shares of Series B preferred stock outstanding which may be converted
into 40,525,000 shares of the Company’s common stock and 851,400 shares common stock to be issued for an aggregate of 41,376,400
shares of common stock.
Research and Development Cost
Research and development (R&D) costs are expensed
as incurred. R&D costs are related to the Company’s internally funded development of the Company medical licenses and patents. The
Company had R&D costs were $-0- for the three and years ended December 31, 2023 and 2022, respectively.
Stock Compensation
The Company accounts for share-based compensation
in accordance with the fair value recognition provisions of the Financial Accounting Standards Board (“FASB”) Accounting Standards
Codification (“ASC”) No. 718 and No. 505. The Company issues restricted stock to employees and consultants for their services.
Cost for these transactions are measured at the fair value of the equity instruments issued at the date of grant. These shares are considered
fully vested and the fair market value is recognized as an expense in the period granted. The Company recognized consulting expenses and
a corresponding increase to additional paid-in-capital related to stock issued for services. For agreements requiring future services,
the consulting expense is to be recognized ratably over the requisite service period.
The Company uses the Black-Scholes-Merton valuation
model for estimating the fair value of traded options and stock warrants. There were no stock warrants or stock options outstanding
on December 31, 2023 and 2022.
The Company recorded stock-based compensation
of $-0- and $1,400 for the years ended December 31, 2023, and 2022, respectively.
Related Parties
The registrant follows subtopic 850-10 of the
FASB Accounting Standards Codification for the identification of related parties and disclosure of related party transactions.
Pursuant to Section 850-10-20 the Related parties
include (a) affiliates of the registrant; (b) entities for which investments in their equity securities would be required, absent the
election of the fair value option under the Fair Value Option Subsection of Section 825–10–15, to be accounted for by the
equity method by the investing entity; (c) trusts for the benefit of employees, such as pension and profit-sharing trusts that are managed
by or under the trusteeship of management; (d) principal owners of the registrant; (e) management of the registrant; (f) other parties
with which the registrant may deal if one party controls or can significantly influence the management or operating policies of the other
to an extent that one of the transacting parties might be prevented from fully pursuing its own separate interests; and (g) other parties
that can significantly influence the management or operating policies of the transacting parties or that have an ownership interest in
one of the transacting parties and can significantly influence the other to an extent that one or more of the transacting parties might
be prevented from fully pursuing its own separate interests.
The financial statements shall include disclosures
of material related party transactions, other than compensation arrangements, expense allowances, and other similar items in the ordinary
course of business. However, disclosure of transactions that are eliminated in the preparation of consolidated or combined financial statements
is not required in those statements. The disclosures shall include: (a) the nature of the relationship(s) involved; (b) description of
the transactions, including transactions to which no amounts or nominal amounts were ascribed, for each of the periods for which income
statements are presented, and such other information deemed necessary to an understanding of the effects of the transactions on the financial
statements; (c) the dollar amounts of transactions for each of the periods for which income statements are presented and the effects of
any change in the method of establishing the terms from that used in the preceding period; and (d) amounts due from or to related parties
as of the date of each balance sheet presented and, if not otherwise apparent, the terms and manner of settlement.
Recently Issued Accounting Standards
Management believes recently issued accounting
pronouncements will have no impact on the financial statements of the Company.
NOTE 2 – GOING CONCERN
The accompanying consolidated financial statements
have been prepared on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities in the normal
course of business. The Company has incurred material recurring losses from operations. The Company has not generated material revenues
since inception and has generated losses totaling $3,923,677 since inception.
The consolidated financial statements do not contain
any adjustments to reflect the possible future effects on the classification of assets or the amounts and classification of liability
that may result should the Company be unable to continue as a going concern.
NOTE 3 – SEGMENT DATA
The Company has three reportable
segments, which it believes best reflect how the Company is currently managed — Generic Drugs, Gene Therapy and Pharmaceutical
Programs. The Generic Drugs segment consists of a portfolio of drugs and biosimilar biologics selling hard-to-source, difficult to find
generic drugs and off-patent biologic therapies and proprietary and patented technology platforms that include a library of melanocortin
receptor-binding molecules, an invitro diagnostic for neurodegenerative diseases. The Generic Drug segment operations focuses on bringing
various generic drugs to market primarily in the U.S. and Europe markets. The Gene Therapy segment uses a gene therapy approach to
introduce a therapeutic protein called “Klotho” inside the body to treat neurodegenerative diseases and other diseases of
aging. The Pharmaceutical Programs segment consists of treatments using small drug molecules that bind to the melanocortin receptors on
human cells and affect skin pigmentation and other initiatives. The assets of the segments consist of the following at December 31, 2023
and 2022:
| | |
December 31, 2023 | | |
December 31, 2022 | |
| | |
| | |
| |
| Generic drugs: | |
| | |
| |
| Licenses | |
$ | 2,101,713 | | |
$ | 2,123,750 | |
| Gene therapy: | |
| | | |
| | |
| Patents | |
| 48,420 | | |
| 86,160 | |
| Pharmaceutical programs: | |
| | | |
| | |
| Licenses | |
| 36,015 | | |
| - | |
| Total | |
$ | 2,186,148 | | |
$ | 2,209,910 | |
The following
table presents the Company’s reportable segment results for the three and Years ended December 31, 2023 and 2022:
| | |
For the Twelve Months Ended | |
| | |
December 31, 2023 | | |
December 31, 2022 | |
| | |
| | |
| |
| Expenses: | |
| | | |
| | |
| Generic drugs | |
$ | 355,358 | | |
$ | 402,543 | |
| Gene therapy | |
| 339,600 | | |
| 196,050 | |
| Pharmaceutical programs | |
| 12,500 | | |
| - | |
| Total | |
| 707,458 | | |
| 598,593 | |
| | |
| | | |
| | |
| Net loss: | |
| | | |
| | |
| Generic drugs | |
| (355,358 | ) | |
| (402,543 | ) |
| Gene therapy | |
| (339,600 | ) | |
| (196,050 | ) |
| Pharmaceutical programs | |
| (12,500 | ) | |
| - | |
| Total | |
$ | (712,095 | ) | |
$ | (598,593 | ) |
NOTE 4 – LICENCES AND PATENTS
Licenses
During 2015, the Company acquired two licenses
two licensed platform technologies, a biosimilar biologics platform that uses biologic therapies to treat cancer – recombinant antibodies,
and a gene therapy platform which uses a gene therapy approach to introduce a therapeutic protein called “Klotho” inside the
body to treat neurodegenerative diseases. The licenses were valued at $736,983.
On September 12, 2022, the Company acquired
four market-approved anti-cancer drugs approved for sale in Germany for $1,308,270. The purchase price consisted of a short-term
promissory note for $1,308,270. The purchase price represents the fair value of the intangible asset based on the net present value of
the projected gross profit to be generated by the licenses.
On January 27, 2023, the Company signed a License
Agreement with Teleost Biopharmaceutic, LLC to acquire various assets for the Company’s proprietary pharmaceutical program segment.
The license includes the use of patented small drug molecules that bind to the melanocortin receptors on human cells and affect skin pigmentation.
The terms include a $10,000 fee for signing the agreement and a $50,000 payment on January 27, 2024. The Company will pay for all new
patent costs for new discoveries and new treatments. The Company will make standard commercial development-based milestone payments for
the various stages of license development and regulatory approval. In addition, the Company will make royalty payments on the net sales
for commercial products. Beginning in 2025, the Company will also pay patent and license maintenance fees.
On March 5, 2023, the Company signed a Non-Exclusive
License Agreement with Heidelberg University to grant non-exclusive rights to various licenses owned and under development by the university.
The licenses includes the use of modified AAV capsid polypeptides for treatment of muscular diseases. The terms include a €50,000
($56,325) fee for signing the agreement and €100,000 ($112,650) payment within 60 days of the anniversary of signing the agreement.
The Company will pay €1,000,000 ($1,126,500) for each assignment of a right to a license owned by the university. For new licenses,
the Company will make standard commercial development-based milestone payments for the various stages of license development and regulatory
approval. The Company will make 2 % royalty payments by January 31st each year during the term of the agreement for each licensed
product for the proceeding calendar year. At December 31, 2023, the Company paid $56,325 under the agreement.
On December 1, 2023, the Company signed a license agreement with TransferTech
Sherbooke for the rights to develop and commercialize the technology of a “Needleless Syringe”. Under the terms of the agreement
the Company pays a $26,060 upfront fee and royalty fees on the license income. In addition, TransferTech Sherbooke will be compensated
with $50,000 in unregistered shares of the Company’s common stock the day prior to the merger pending merger with Redwoods Acquisition
Corp. The shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split
of its common stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The Company
has not commenced developing the technology. The amount due under the agreement was $26,060 at December 31, 2023.
The total licenses were $2,137,638 and $2,123,750
at December 31, 2023 and 2022, respectively, in the accompanying consolidated balance sheet. The licenses are not in use. Once the licenses
are in use, the licenses will be amortized over the useful life.
Patents
The Company is acquiring patents for Alzheimer,
ALS and other items from third-party. As of December 31, 2023, the patents have not been finalized. Once the patents are declared effective,
the Patents will be amortized over the shorter of a patent’s useful life and its legal useful
life. The patent cost incurred as of December 31, 2023 and 2022 was $48,420 and $86,160, respectively, and reported as patents in the
accompanying consolidated balance sheet. At December 31, 2023, certain patent costs of $47,740 was deemed not capitalizable and were expensed
as professional fees in the accompanying statements for operations.
NOTE 5 – NOTES PAYABLE
On September 12, 2022, the Company issued a $1,308,270
promissory note to acquired four market-approved anti-cancer drugs. See Note 4 – Licenses and Patents for a further
discussion. The promissory note bears interest at 6% and a maturity date of June 30, 2023. The Company has agreed to make a monthly interest
payment of $6,541. By agreement, the interest will stop accruing at June 30, 2023. As of December 31, 2023 and Company made interest payments
of $78,496 to fully satisfy the interest obligation under the promissory note. The unpaid balance principal balance was $1,308,270 and
$1,352,260 at December 31, 2023 and 2022, respectively.
On December 12, 2023, the Company issued a $24,000
promissory note to a member of the CEO household and not a related party. The promissory note accrued interest at a one-time interest
fee of $2,460. The unpaid balance principal and interest balance was $26,460 at December 31, 2023.
NOTE 6 – EQUITY TRANSACTIONS
The Company was established with three classes
of stock, common stock – 1,500,000,000 shares authorized at a par value of $0.0001 Class B preferred stock 500,000 shares authorized
at a par value of $0.001 and Class C preferred stock 5,000,000 shares authorized at a par value of $0.0001.
On December 31, 2023 and 2022, the Company issue
and outstanding common stock was 1,044,861,360 shares, 405,250 shares of Class B preferred stock and 1,000,000 shares of Class C preferred
stock.
On November 1, 2021, the shareholders of the Company
approved a 1-for-2500 reverse split. As of December 31, 2023, the reverse split has not been declared effective by FINRA to broker deals
in the quotation system.
On February 1, 2022, the Company entered into
a Stock Purchase Agreement with an individual to sell 1,666,667 shares of the Company’s common stock for $250,000 or $0.15 shares. The
shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split of its common
stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The shares have not been
issued to the individual at December 31, 2023.
On February 22, 2022, the Company entered into
a consulting agreement to provide service to the Company. Pursuant to the agreement, the consultant is compensated with 1,000,000 shares
of the Company’s restricted common stock. The shares were valued at $0.0014 per share. The shares have not been issued to the consultant
on December 31, 2023.
On September 12, 2022, the Company entered into
a Stock Purchase Agreement with an individual to sell 2,000,000 shares of the Company’s common stock for $500,000 or $0.25 shares. The
shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split of its common
stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The shares have not been
issued to the individual at December 31, 2023.
On August 15, 2023, the Company entered into a
Stock Purchase Agreement with an individual to sell 300,000 shares of the Company’s restricted common stock for $75,000 or $0.25
shares. The shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split
of its common stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The shares have
not been issued to the individual on December 31, 2023.
On August 15, 2023, the Company entered into a Stock Purchase Agreement
with an individual to sell 100,000 shares of the Company’s restricted common stock for $25,000 or $0.25 shares. The shares issued
will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split of its common stock was
not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The shares have not been issued to the
individual on December 31, 2023.
NOTE 7 – MATERIAL CONTRACTS
On November 27, 2014, the Company signed a License
Agreement and a Manufacturing and Supply Agreement for the monoclonal antibody development license and supply agreement and related
manufacturing with Reliance Life Sciences (RLS), the life science arm of Reliance Industries Pvt Ltd, the largest private company in India.
The contract expires on November 27, 2024 with a 10-year renewal option. The License Agreement entitles the Company to pay $100,000 per
product for a total of three products with milestone payments for meeting certain criteria. In addition, the Company will pay a quarterly
royalty payment of 5% on net sales of finished products. The Manufacturing and Supply Agreement contains an estimated acquisition price
of active pharmaceutical ingredients (API ) of $350,000 per Kg for each product developed. As of December 31, 2023, the Company has
not generated any activity under the agreement.
On October 1, 2020, the Company entered into a
three-year Management Consulting Services Agreement with an individual to provide various services including raising funds for the Company.
The contact terminates on December 31, 2023. The consultant is compensated with 3% of the net proceeds of the any contractual relationship
and equity compensation of up to 3% of the value of the business development contract with restricted share of the Company’s common
stock. As of December 31, 2023, the Company has not generated any activity under the agreement.
On November 19, 2021, the Company sign a consulting
agreement with an individual to raise capital for new medical products and commercialize such products for a 5% commission fee. As of
December 31, 2023, the Company has not generated any activity under the agreement.
On January 24, 2022, the Company signed an exclusive,
world-wide License Agreement with the University of Barcelona for a cell and/or gene therapy that has shown compelling activity in animal
models of human Alzheimer’s disease and amyotrophic lateral sclerosis (“ALS” or “Lou Gehrig’s disease”).
The gene therapy will also be applied to age-related diseases and rare (“Orphan”) diseases. Beginning on December 15, 2022,
the Quarterly license fee is 10,000 Euros. In addition, the Company will pay a Royalty equal to 3% of net sales of finished products.
For the years ended December 31, 2023 and 2022, the Company owes $-0- under the agreement.
On April 5, 2022, the Company entered into a Business
Development and Consulting Agreement with an individual to serve as the Company’s chief business officer. Beginning on May 1, 2022,
the consultant is compensated with $10,000 a month for the three months ended July 31, 2022 and $15,000 thereafter. The consultant works
approximately 80 hours a month. In addition to cash considerations, the consultant was compensated with 1,000,000 shares of the Company’s
common stock valued at $1,400 or $0.0014 per share. As of December 31, 2023, the shares have not been issued to the consultant. The Contract
was terminated on August 15, 2022 with no amount due to the consultant.
On October 19, 2022, the Company sign a M&A/Capital
Markets Advisory Agreement with a firm to advise and assist the Company in negotiating the terms and conditions with respect to a potential
sale, purchase, merger, joint venture, business combination, material change of control, or similar transaction involving the Company
and a strategic acquirer and/or private or publicly listed entity or business, including a Special Purpose Acquisition Company (SPAC),
and with respect to any offerings of any equity, equity-linked or debt securities of the Company or any other party to a financing transaction
and perform such other financial advisory services to the Company. The Company will compensate the firm with an M&A fee, a financing
fee and expenses.
Upon consummation of a transaction, the Company
will pay the firm an M&A fee consisting of an aggregate of a sum equal to the greater of $2,500,000 or the sum of the following amounts:
| ● | four percent (4.0%) of the first $100 MM of Aggregate Value; |
| ● | three percent (3.0%) of any amount of the Aggregate Value between
$100 MM and $200 MM; |
| ● | two percent (2.0%) of any amount of the Aggregate Value between
$200 MM and $300MM; |
| ● | one percent (1.0%) of any amount of the Aggregate Value exceeding
$300 MM |
In addition, the Company will pay the firm a financing
fee of seven percent (7%) of the aggregate amount of proceeds received from investors in the financing of any equity or equity-linked
securities and three percent (3%) of the aggregate amount of proceeds received from the Financing of any non-equity-linked debt securities
and credit facilities. As of December 31, 2023, the Company owes $-0- under the agreement.
On February 1, 2023, the Company entered into
a Consulting Agreement with an individual to advise the Company in the general field of melanocortins, melanocortin receptors and melanocortin
receptor-binding molecules. The consultant will be compensated with $2,500 for 12 months ending on February January 31, 2024. The Consultant
was paid $10,000 upfront for the first four months and $2,500 during March 2023 for an aggregate of $12,500. The Contract was terminated
during March 2023 and with no amount due to or due from the Consultant.
NOTE 8 – RELATED PARTIES
On June 8, 2020, the Company signed a Management
Consulting Services Agreement with an individual to provide services to the Company. In addition, the individual has been appointed a
director and an officer of the Company. The individual is compensated with $10,000 per month. At December 31, 2023, the individual
is owed $55,000 under the consulting agreement with the Company.
On October 10, 2021, the Company signed an Employment
Agreement with Dr, Joseph Sinkule to serve as the Company’s CEO for three years ending on October 9th 2024. In addition,
My Sinkule will serve as a member of the board of directors for a five-year term. Mr. Sinkule’s annual salary will be $240,000 per
year and increase to $360,000 per year upon raising a total of five million dollars ($5,000,000) or more in equity and/or debt financing.
The Company’s CEO has earned $240,000 for the years ended December 31, 2023 and 2022. At December 31, 2023, the Company’s
CEO is owed $80,000 under the agreement.
During November 2022, the Company advanced a
shareholder $300,000 as a short-term loan. The loan is non-interest bearing and due by the end of December 2022. The shareholder repaid
$50,000 during December 2022 and $250,000 in January 2023 to fully satisfy the advance. At December 31, 2023 and 2022, the loan balance
was $-0- and $250,000, respectively and is reported in due from related party in the accompanying consolidated balance sheets.
At December 31, 2023, the aggregate related party
payable was $135,000 and is reported as related party payable in the accompanying consolidated balance sheets.
NOTE 9 – INCOME TAXES
The Company’s policy is to provide for deferred income taxes
based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates that will be in
effect when the differences are expected to reverse. We did not provide any current or deferred U.S. federal income tax provision or benefit
for any of the periods presented because we have experienced operating losses since inception. When it is more likely than not that a
tax asset cannot be realized through future income the Company must allow for this future tax benefit. We provided a full valuation allowance
on the net deferred tax asset, consisting of net operating loss carryforwards, because management has determined that it is more likely
than not that we will not earn income sufficient to realize the deferred tax assets during the carryforward period.
The Company is not aware of any uncertain tax
position that, if challenged, would have a material effect on the financial statements for the year ended December 31, 2023 or during
the prior three years applicable under FASB ASC 740. We did not recognize any adjustment to the liability for uncertain tax position and
therefore did not record any adjustment to the beginning balance of accumulated deficit on the balance sheet. All tax returns for the
Company remain open for examination.
The provision for income taxes differs from the
amount computed by applying the statutory federal income tax rate to income before provision for income taxes. The sources and tax effects
of the differences for the periods presented are as follows:
| | |
2023 | | |
2022 | |
| Income tax provision at the federal statutory rate | |
| 28 | % | |
| 21 | % |
| Effect on operating losses | |
| (28 | )% | |
| (21 | )% |
The net deferred tax assets consist of the following:
| | |
December 31, 2023 | | |
December 31, 2022 | |
| Deferred tax asset | |
$ | 1,098,630 | | |
$ | 675,406 | |
| Valuation allowance | |
| (1,0968,630 | ) | |
| (675,406 | ) |
| Net deferred tax asset | |
$ | - | | |
$ | - | |
The change in the valuation allowance for the
year ended December 31, 2022 was an increase of $423,224.
NOTE 10 – SUBSEQUENT EVENTS
On January 19, 2024, the Company entered into
a Stock Purchase Agreement with an individual to sell 200,000 shares of the Company’s restricted common stock for $50,000 or $0.25
shares. The shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split
of its common stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The shares have
not been issued to the individual on May 22, 2024.
The Company’s S4 with Redwoods Acquisition
Corp. was declared effective by the SEC on February 14, 2024. The required approval by the stockholders of Redwoods and the Company, approval
by the Nasdaq Stock Market (“Nasdaq”) of the initial listing application of the combined company filed in connection with
the Business Combination, and the fulfillment of other customary closing conditions is expected to be completed in Mayl 2024.
On February 29, 2024, the Company granted three
individuals, two executives and an organization 9,900,000 unregistered shares of the Company’s Common stock for services performed
for the Company. The shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse
stock split of its common stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The
shares were valued at $43,560 or $0.0044 per share. The shares have not been issued to the individuals, executives or the university on
May 22, 2024.
On March 1, 2024, the Company filed a Certificate
of Designation with the State of Wyoming to authorize 160,000 shares of Series D Preferred Stock with a par value of $0.001. The Series
D Preferred Stock shall earn cumulative cash dividend at the annual rate of 6.0% of the original purchase price per share. Each Series
D Preferred Stock shall be converted into such number of fully paid and nonassessable shares of common stock as determined by (i) dividing
the original issue price by the automatic conversion rate and (ii) and dividing by the result quotient of ten (10). Each share of Series
D Preferred Stock shall automatically be converted into shares of the Company stock prior to the consummation of the Company’s SPAC
transaction.
On March 4, 2024, the Company sold 50,000 shares
of Series D Preferred Stock for $125,000 or $2.5 per share to an investor. In addition, the investor was granted 100,000 shares of =Series
D Preferred Stock as a commitment fee and a warrant to purchase 1,500,000 shares of the Company’s unregistered common stock. The
warrant has an excise price of $2.5 per share and expires on March 4, 2027. The Company will value the warrant using the Black-Scholes-Merton
valuation model for estimating the fair value of call options.
On March 15, 2024, the Company signed a Management
Consulting Services Agreement with an individual in exchange of 30,000 shares of Series B Preferred stock originally issued on November
1, 2021. On March 31, 2024, the Company cancelled the 30,000 shares of Series B Preferred Stock. The individual is granted a call option
for 3,000,000 unregistered shares of the Company’s Common stock at a $30 acquisition price. The call option expires on March 15,
2029. The shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split
of its common stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The shares were
valued at $18,000 or $0.006 per share. The shares have not been issued to the individuals, executives or the university on May 22, 2024.
On March 15, 2024, the Company signed a Management
Consulting Services Agreement to an individual for services to the Company. The individual is compensated with a call option for 1,000,000
unregistered shares of the Company’s Common stock at a $10 acquisition price. The call option expires on March 15, 2029. The shares
issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse stock split of its common stock
was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The shares were valued at $6,000
or $0.006 per share. The shares have not been issued to the individuals, executives or the university on May 22, 2024.
On March 15, 2024, the Company signed a Management
Consulting Services Agreement with a university for services to the Company. The university is compensated with a call option for 2,000,000
unregistered shares of the Company’s Common stock at a $20 acquisition price. The call option expires on March 15, 2029. The Company
has a planned 1-for-2500 reverse stock split of its common stock was not declared effective as of December 31, 2023 by FINRA to broker
deals in the quotation system. The shares were valued at $12,000 or $0.006 per share. The shares have not been issued to the individuals,
executives or the university on May 22, 2024.
On March 25, 2024, the Company signed an Equity
for Consulting Agreement with an individual to serve as the Company’s Chief Business Officer. The individual is compensated with
1,000,000 unregistered shares of the Company’s Common stock and $10,000 per month effective the first day of Company’s de-SPAC
and public trading. The shares issued will be calculated on a post-reverse split basis. The Company has a planned 1-for-2500 reverse
stock split of its common stock was not declared effective as of December 31, 2023 by FINRA to broker deals in the quotation system. The
shares were valued at $6,700 or $.0067 per share. The shares have not been issued to the individual on May 22, 2024.
On March 27, 2024, the Company amended the December 1, 2023 license
agreement with TransferTech Sherbooke to remove the equity compensation of $50,000 in unregistered shares of the Company’s common
stock for a lump sum cash payment of $50,000 within 10 days after the pending merger with Redwoods Acquisition Corp. The December 1, 2023
license agreement with TransferTech Sherbooke contains the rights to develop and commercialize the technology of a “Needleless Syringe”.
On March 31, 2024, the Company cancelled 50,000 authorized but unissued unregistered shares of the Company’s Common stock which
were due under the agreement.
On March 28, 2024, the Company signed a Securities Purchase Agreement
with an investor for a $1,300,000 convertible promissory note which will fund upon closing the business combination with Redwoods Acquisition
Corp under the Company’s S-4 declared effective on February 14, 2024. The business combination is expected to close in May 2024.
This convertible promissory note will mature 24 months after the aforementioned closing date. The interest on this note shall commence
accruing on the original issuance date and shall be payable, at the Company’s
option, either (i) in cash at 10% per annum or (ii) in freely tradable common shares (at the lower of the conversion price or 10%
discount to the lowest of 5-day VWAP prior to the interest payment date) at 15%
per annum. In addition, the convertible promissory note may be converted upon the aforementioned closing of the business combination
at $9.00 per share; provided, however, the conversion price shall be subject
to a conversion reset as set forth in this convertible note payable. The floor conversion price is the floor price that is in compliance
with the requirements of the Nasdaq Stock Market LLC or another national exchange.
On March 28, 2024, the Company signed a Securities Purchase Agreement
with an investor for a $700,000 convertible promissory note which will fund upon closing the business combination with Redwoods Acquisition
Corp under the Company’s S-4 declared effective on February 14, 2024. The business combination is expected to close in May 2024.
This convertible promissory note will mature 24 months after the aforementioned closing date. The interest on this note shall commence
accruing on the original issuance date and shall be payable, at the Company’s
option, either (i) in cash at 10% per annum or (ii) in freely tradable common shares (at the lower of the conversion price or 10%
discount to the lowest of 5-day VWAP prior to the interest payment date) at 15%
per annum. In addition, the convertible promissory note may be converted upon the aforementioned closing of the business combination
at $9.00 per share; provided, however, the conversion price shall be subject
to a conversion reset as set forth in this convertible note payable. The floor conversion price is the floor price that is in compliance
with the requirements of the Nasdaq Stock Market LLC or another national exchange.
On April 22, 2024, the Company signed a $2,000,000 convertible promissory
note with an investor. The convertible promissory note will mature on April 22, 2026. The note is expected to fund upon closing the business
combination with Redwoods Acquisition Corp under the Company’s S-4 declared effective on February 14, 2024. The business combination
is expected to close in May 2024.The interest on this note shall be payable, at
the Company’s option, either (i) in cash at 10% per annum or (ii) in freely tradable Common Shares (at the lower of the conversion
price or 10% discount to the lowest of 5-day VWAP prior to the interest payment date) at
15% per annum. In addition, the convertible promissory note may be converted upon the aforementioned closing of the business combination
at $9.00 per share; provided, however, the conversion price shall be subject
to a conversion reset as set forth in this convertible note payable. The floor conversion price is the floor price that is in compliance
with the requirements of the Nasdaq Stock Market LLC or another national exchange.
The Company evaluated all events or transactions
that occurred through May 22, 2024. During this period, the Company did not have any other material recognizable subsequent events.
Up
to a Maximum of 21,527,925 Shares of Common Stock
at $0.95 per Share
Preliminary
Prospectus
ANEW
MEDICAL, INC.
September
5, 2024
YOU
SHOULD ONLY RELY ON THE INFORMATION CONTAINED IN THIS PROSPECTUS. WE HAVE NOT AUTHORIZED ANYONE TO PROVIDE YOU WITH INFORMATION
DIFFERENT FROM THAT CONTAINED IN THIS PROSPECTUS. THE SELLING STOCKHOLDERS ARE OFFERING TO SELL, AND SEEKING OFFERS TO BUY, COMMON SHARES
ONLY IN JURISDICTIONS WHERE OFFERS AND SALES ARE PERMITTED.
PART
II - INFORMATION NOT REQUIRED IN PROSPECTUS
Item
13. Other Expenses of Issuance and Distribution
The
following table indicates the expenses to be incurred in connection with this offering, other than the placement agent fees, all of which
will be paid by us. All amounts are estimated except the Securities and Exchange Commission registration fee and the Financial Industry
Regulatory Authority, Inc. (“FINRA”) filing.
| | |
Amount | |
| SEC registration
fee | |
$ | 3,369 | |
| Accountants’ fees
and expenses | |
$ | 5,000 | |
| Legal fees and expenses | |
$ | 20,000 | |
| Miscellaneous | |
$ | 3,075 | |
| Total expenses | |
$ | 31,444 | |
Item
14. Indemnification of Directors and Officers
We
shall indemnify any officer or director or any former officer or director, to the full extent permitted by law. We shall indemnify
any officer or director in connection with any proceedings, including appeals, if he or she acted in good faith and in a manner he or
she reasonably believed to be in our best interests and they had no reasonable cause to believe that his or her conduct was unlawful.
The termination of any proceeding by judgment, order, settlement, or conviction or upon a plea of nolo contendere or its equivalent
shall not, of itself, create a presumption that the person did not act in good faith and in a manner which he or she reasonably believed
to be in our best interests or had reasonable cause to believe that his or her conduct was unlawful.
At
present, there is no pending litigation or proceeding involving any of our directors or executive officers as to which indemnification
is required or permitted, and we are not aware of any threatened litigation or preceding that may result in a claim for indemnification.
Item
15. Recent Sales of Unregistered Securities
There
were no unregistered securities to report which have not been previously included in a Quarterly Report on Form 10-Q or a Current Report
on Form 8-K.
Item
16. Exhibits and Financial Statement Schedules
(a)
Exhibits.
The
following exhibits are filed as part of this registration statement:
| Exhibit No. |
|
Description |
| 2.1 |
|
Business
Combination Agreement, dated May 30, 2023, by and among Redwoods Acquisition Corp., ANEW MEDICAL SUB, INC. and ANEW MEDICAL,
INC. ** |
| 2.2 |
|
Amendment
No.1 to Business Combination Agreement, dated November 4, 2023, by and among Redwoods Acquisition Corp., ANEW MEDICAL SUB, INC. and
ANEW MEDICAL, INC. ** |
| 3.1 |
|
Amended
and Restated Certificate of Incorporation of Redwoods Acquisition Corp. (incorporated by reference to Exhibit 3.1 to Redwoods’
Current Report on Form 8-K filed with the SEC on April 4, 2022).** |
| 3.2 |
|
Bylaws
of Redwoods Acquisition Corp. (incorporated by reference to Exhibit 3.4 filed with Redwoods’ registration statement on Form S-1
filed by the Registrant on March 10, 2022).** |
| 3.3 |
|
Form
of Second Amended and Restated Certificate of Incorporation of Redwoods Acquisition Corp. ** |
| 3.4 |
|
Form
of Amended and Restated Bylaws of Redwoods Acquisition Corp. ** |
| 4.1 |
|
Specimen
Unit Certificate (incorporated by reference to Exhibit 4.1 to Redwoods’ Registration Statement on Form S-1 filed
with the SEC on March 10, 2022).** |
| 4.2 |
|
Specimen
Class A Common Stock Certificate. ** |
| 4.3 |
|
Specimen
Warrant Certificate ** |
| 4.4 |
|
Warrant
Agreement between Continental Stock Transfer & Trust Company and the Registrant ** |
| 5.1 |
|
Legal opinion of Cyruli Shanks & Zizmor, LLP |
| 10.1 |
|
Lock-Up
Agreement, dated as of May 30, 2023, by and among Redwoods Acquisition Corp. and the other parties named therein ** |
| 10.2 |
|
Sponsor
Support Agreement, dated as of December 29, 2023, by and among Redwoods Acquisition Corp., Redwoods Capital LLC, and other parties
thereto ** |
| 10.3 |
|
Voting
and Support Agreement, dated as of May 30, 2023, by and among Redwoods Acquisition Corp., ANEW MEDICAL, INC. and the other parties
named therein ** |
| 10.4 |
|
ANEW
MEDICAL, INC. 2023 Stock Incentive Plan ** |
| 10.5 |
|
Registration
Rights Agreement, dated May 30, 2023, by and among Redwoods Acquisition Corp., certain stockholders of ANEW MEDICAL, INC. and the
Founder Holders (incorporated by reference to Exhibit 10.3 to Redwoods’ Current Report on Form 8-K filed with the SEC on June
5, 2023).** |
| 10.6 |
|
Letter
Agreement, dated March 30, 2022, by and among Redwoods Acquisition Corp. and its officers, directors and the Sponsor (incorporated
by reference to Exhibit 10.1 to Redwoods’ Current Report on Form 8-K filed with the SEC on April 4, 2022).** |
| 10.7 |
|
Investment
Management Trust Agreement, dated March 30, 2022, by and between Redwoods Acquisition Corp. and Continental Stock Transfer &
Trust Company, LLC (incorporated by reference to Exhibit 10.2 to Redwoods’ Current Report on Form 8-K filed with the SEC
on April 4, 2022).** |
| 10.8 |
|
Registration
Rights Agreement, dated March 30, 2022, by and among Redwoods Acquisition Corp. and certain security holders named therein (incorporated
by reference to Exhibit 10.4 to Redwoods’ Current Report on Form 8-K filed with the SEC on April 4, 2022).** |
| 10.9 |
|
Administrative
Support Agreement, dated March 30, 2022, by and between Redwoods Acquisition Corp. and the Sponsor (incorporated by reference to
Exhibit 10.8 to Redwoods’ Current Report on Form 8-K filed with the SEC on April 4, 2022).** |
| 10.10 |
|
Indemnity
Agreements, Each dated as of March 30, 2022, by and between the Registrant and Each of the officers and directors of the Registrant
(incorporated by reference to Exhibit 10.7 to Redwoods’ Current Report on Form 8-K filed with the SEC on April 4,
2022).** |
| 10.11 |
|
Subscription
Agreement, dated March 30, 2022, by and between the Registrant and the Sponsor (incorporated by reference to Exhibit 10.5 to Redwoods’
Current Report on Form 8-K filed with the SEC on April 4, 2022).** |
| 10.12 |
|
Subscription
Agreement, dated March 30, 2022, by and between the Registrant and the Sponsor (incorporated by reference to Exhibit 10.6 to Redwoods’
Current Report on Form 8-K filed with the SEC on April 4, 2022).** |
| 10.13 |
|
Sponsored
Research Agreement with Universitat Autònoma de Barcelona.** |
| 10.14 |
|
Exclusive
license with Universitat Autònoma de Barcelona and Institució Catalana De Recerca I Estudis Avançats for the
cognition and Alzheimer’s related inventions covered by USPTO Application No.: 15/777,456.** |
| 10.15 |
|
Exclusive
license with Universitat Autònoma de Barcelona and Institució Catalana De Recerca I Estudis Avançats for the
neuromuscular related inventions covered by USPTO Application No.: 18/299,989.** |
| 10.16 |
|
Non-exclusive
license with University of Heidelberg, Germany for the myotropic AAV capsids related inventions covered by USPTO Application No.
17/051,123.** |
| 10.17 |
|
Exclusive
license agreement and exclusive license and manufacturing agreement in licensed territories with Reliance Life Science Private Limited.** |
| 10.18 |
|
Exclusive
sublicense license agreement with Teleost Biopharmaceuticals, LLC (AZ) for the melanocortins related technologies covered by US Patent
9,441,013, US Patent 10,329,326, US Patent 9,290,539, US Patent 9,539,301 and European Patent 3,177,737.** |
| 14 |
|
Code
of Ethics of Redwoods Acquisition Corp. (incorporated by reference to Exhibit 14 to Redwoods’ Registration Statement on
Form S-1 filed with the SEC on March 10, 2022).** |
| 23.1 |
|
Consent of Cyruli Shanks & Zizmor, LLP (included in Exhibit 5.1) |
| 23.2 |
|
Consent of Yusufali & Associates, LLP |
| 101.INS |
|
XBRL
Instance Document |
| 101.SCH |
|
XBRL
Taxonomy Extension Schema Document |
| 101.CAL |
|
XBRL
Taxonomy Extension Calculation Linkbase Document |
| 101.DEF |
|
XBRL
Taxonomy Extension Definition Linkbase Document |
| 101.LAB |
|
XBRL
Taxonomy Extension Label Linkbase Document |
| 101.PRE |
|
XBRL
Taxonomy Extension Presentation Linkbase Document |
| 104 |
|
Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
| 107 |
|
Filing Fee Calculation Table |
| (b) | Financial
Statement Schedules. |
All
financial statement schedules have been omitted, since the required information is not applicable or is not present in amounts sufficient
to require submission of the schedule, or because the information required is included in the financial statements and notes thereto.
Item
17. Undertakings
(a)
The undersigned registrant hereby undertakes:
(1)
To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
i.
To include any prospectus required by Section 10(a) (3) of the Securities Act of 1933;
ii.
To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective
amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration
statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of
securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering
range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes
in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of
Registration Fee” table in the effective registration statement.
iii.
To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or
any material change to such information in the registration statement.
(2)
That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed
to be a new registration statement relating to the securities offered, and the offering of such securities at that time shall be deemed
to be the initial bona fide offering thereof.
(3)
To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the
termination of the offering.
(4)
That, for the purpose of determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b)
as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses
filed in reliance on Rule 430A (§ 230.430A of Title 17 of the Code of Federal Regulations), shall be deemed to be part of and included
in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration
statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference
into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract
of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was
part of the registration statement or made in any such document immediately prior to such date of first use.
(5)
That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution
of the securities:
The
undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration
statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold
to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will
be considered to offer or sell such securities to such purchaser:
(i)
Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule
424 (§230.424 of Title 17 of the Code of Federal Regulations);
(ii)
Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by
the undersigned registrant;
(iii)
The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant
or its securities provided by or on behalf of the undersigned registrant; and
(iv)
Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
Insofar
as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons
of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities
and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the
event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid
by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted
by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the
opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question
whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of
such issue.
SIGNATURES
Pursuant
to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf
by the undersigned, thereunto duly authorized in the City of Omaha, State of Nebraska, on September 5,
2024.
| ANEW Medical, Inc. |
|
| |
|
|
| By: |
/s/ Joseph A. Sinkule |
|
| |
Joseph A. Sinkule |
|
| |
Chief Executive Officer |
|
Pursuant
to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities
and on the dates indicated.
POWER
OF ATTORNEY
KNOW
ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Joseph A, Sinkule, as his true
and lawful attorneys-in-fact and agents, with full powers of substitution and resubstitution, for him and in his name, place and stead,
in any and all capacities, to sign any and all amendments to this Registration Statement (including post-effective amendments and any
related registration statements filed pursuant to Rule 462 and otherwise), and to file the same with all exhibits thereto, and other
documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents and full
power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully
for all intents and purposes as he might or could do in person, hereby ratifying and confirming that said attorney-in-fact and agent,
or any substitute or resubstitute, may lawfully do or cause to be done by virtue hereof.
Pursuant
to the requirements of the Securities Act, this registration statement has been signed by the following persons in the capacities and
on the dates indicated.
| Name |
|
Capacity
in Which Signed |
|
Date |
| |
|
|
|
|
| /s/
Joseph A. Sinkule |
|
Chief
Executive Officer and Chairman |
|
September
5, 2024 |
| Dr. Joseph
A. Sinkule |
|
(Principal Executive Officer) |
|
|
| |
|
|
|
|
| /s/
Jeffrey LeBlanc |
|
Chief
Financial Officer |
|
September
5, 2024 |
| Jeffrey
LeBlanc |
|
(Principal Financial and
Accounting officer) |
|
|
| |
|
|
|
|
| /s/
Shalom Z. Hirschman |
|
Director |
|
September
5, 2024 |
| Dr. Shalom
Z. Hirschman |
|
|
|
|
| |
|
|
|
|
| /s/
Samuel Zentman |
|
Director |
|
September
5, 2024 |
| Samuel
Zentman |
|
|
|
|
| |
|
|
|
|
| /s/
Jon W. McGarity |
|
Director |
|
September
5, 2024 |
| Jon W.
McGarity |
|
|
|
|
II-6
0.02
0.03
0.03
0.07
false
0001907223
0001907223
2024-01-01
2024-06-30
0001907223
2024-06-30
0001907223
2023-12-31
0001907223
us-gaap:RelatedPartyMember
2024-06-30
0001907223
us-gaap:RelatedPartyMember
2023-12-31
0001907223
2024-04-01
2024-06-30
0001907223
2023-04-01
2023-06-30
0001907223
2023-01-01
2023-06-30
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:CommonStockMember
2023-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:PreferredStockMember
2023-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:AdditionalPaidInCapitalMember
2023-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
wena:CommonStockToBeIssuedMember
2023-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:RetainedEarningsMember
2023-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
2023-12-31
0001907223
us-gaap:CommonStockMember
2024-01-01
2024-06-30
0001907223
us-gaap:PreferredStockMember
2024-01-01
2024-06-30
0001907223
us-gaap:AdditionalPaidInCapitalMember
2024-01-01
2024-06-30
0001907223
wena:CommonStockToBeIssuedMember
2024-01-01
2024-06-30
0001907223
us-gaap:RetainedEarningsMember
2024-01-01
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:CommonStockMember
2023-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:PreferredStockMember
2023-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:AdditionalPaidInCapitalMember
2023-12-31
0001907223
srt:RestatementAdjustmentMember
wena:CommonStockToBeIssuedMember
2023-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:RetainedEarningsMember
2023-12-31
0001907223
srt:RestatementAdjustmentMember
2023-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:CommonStockMember
2024-01-01
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:PreferredStockMember
2024-01-01
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:AdditionalPaidInCapitalMember
2024-01-01
2024-06-30
0001907223
srt:RestatementAdjustmentMember
wena:CommonStockToBeIssuedMember
2024-01-01
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:RetainedEarningsMember
2024-01-01
2024-06-30
0001907223
srt:RestatementAdjustmentMember
2024-01-01
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:CommonStockMember
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:PreferredStockMember
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:AdditionalPaidInCapitalMember
2024-06-30
0001907223
srt:RestatementAdjustmentMember
wena:CommonStockToBeIssuedMember
2024-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:RetainedEarningsMember
2024-06-30
0001907223
srt:RestatementAdjustmentMember
2024-06-30
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:CommonStockMember
2022-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:PreferredStockMember
2022-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:AdditionalPaidInCapitalMember
2022-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
us-gaap:RetainedEarningsMember
2022-12-31
0001907223
srt:ScenarioPreviouslyReportedMember
2022-12-31
0001907223
us-gaap:CommonStockMember
2023-01-01
2023-06-30
0001907223
us-gaap:PreferredStockMember
2023-01-01
2023-06-30
0001907223
us-gaap:AdditionalPaidInCapitalMember
2023-01-01
2023-06-30
0001907223
us-gaap:RetainedEarningsMember
2023-01-01
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:CommonStockMember
2022-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:PreferredStockMember
2022-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:AdditionalPaidInCapitalMember
2022-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:RetainedEarningsMember
2022-12-31
0001907223
srt:RestatementAdjustmentMember
2022-12-31
0001907223
srt:RestatementAdjustmentMember
us-gaap:CommonStockMember
2023-01-01
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:PreferredStockMember
2023-01-01
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:AdditionalPaidInCapitalMember
2023-01-01
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:RetainedEarningsMember
2023-01-01
2023-06-30
0001907223
srt:RestatementAdjustmentMember
2023-01-01
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:CommonStockMember
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:PreferredStockMember
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:AdditionalPaidInCapitalMember
2023-06-30
0001907223
srt:RestatementAdjustmentMember
us-gaap:RetainedEarningsMember
2023-06-30
0001907223
srt:RestatementAdjustmentMember
2023-06-30
0001907223
2022-12-31
0001907223
2023-06-30
0001907223
wena:RedwoodsAcquisitionCorpMember
2024-01-01
2024-06-30
0001907223
wena:RedwoodsAcquisitionCorpMember
2024-06-30
0001907223
us-gaap:CommonStockMember
2024-06-30
0001907223
wena:PublicWarrantsMember
2024-06-30
0001907223
wena:PrivateWarrantMember
2024-06-30
0001907223
2024-06-21
2024-06-21
0001907223
wena:RedwoodsMember
2024-06-30
0001907223
wena:ANewMedicalIncMember
2024-06-30
0001907223
wena:MergerSubCommonStockMember
2024-06-30
0001907223
2024-03-04
2024-03-04
0001907223
2024-03-04
0001907223
us-gaap:SeriesOfIndividuallyImmaterialBusinessAcquisitionsMember
2024-03-04
0001907223
2024-04-22
2024-04-22
0001907223
2024-04-22
0001907223
us-gaap:SeriesOfIndividuallyImmaterialBusinessAcquisitionsMember
2024-04-22
0001907223
wena:RedwoodsCommonStockMember
2024-06-30
0001907223
wena:ContingentConsiderationSharesMember
2024-06-30
0001907223
2024-06-21
0001907223
us-gaap:CashMember
2024-06-30
0001907223
us-gaap:FairValueInputsLevel1Member
2024-01-01
2024-06-30
0001907223
us-gaap:FairValueInputsLevel2Member
2024-01-01
2024-06-30
0001907223
us-gaap:FairValueInputsLevel3Member
2024-01-01
2024-06-30
0001907223
2023-01-01
2023-12-31
0001907223
us-gaap:FairValueInputsLevel1Member
2023-01-01
2023-12-31
0001907223
us-gaap:FairValueInputsLevel2Member
2023-01-01
2023-12-31
0001907223
us-gaap:FairValueInputsLevel3Member
2023-01-01
2023-12-31
0001907223
2024-03-31
0001907223
2023-03-31
0001907223
us-gaap:WarrantMember
2024-01-01
2024-06-30
0001907223
us-gaap:WarrantMember
2023-01-01
2023-06-30
0001907223
us-gaap:CommonStockMember
2024-06-30
0001907223
wena:DOInsuranceMember
2024-06-30
0001907223
wena:DOInsuranceMember
2023-12-31
0001907223
us-gaap:LicensingAgreementsMember
2024-01-01
2024-06-30
0001907223
2022-09-12
2022-09-12
0001907223
2022-12-15
2022-12-15
0001907223
2024-01-27
2024-01-27
0001907223
2023-03-05
2023-03-05
0001907223
2024-01-31
2024-01-31
0001907223
2024-06-30
2024-06-30
0001907223
2023-12-01
2023-12-01
0001907223
us-gaap:PatentsMember
2023-01-01
2023-12-31
0001907223
us-gaap:PatentsMember
2024-01-01
2024-06-30
0001907223
2014-11-27
2014-11-27
0001907223
2014-11-27
0001907223
wena:RelianceIndustriesPvtLtdMember
2014-11-27
2014-11-27
0001907223
2024-06-16
2024-06-21
0001907223
us-gaap:CommonStockMember
2024-06-21
0001907223
2022-09-12
0001907223
wena:PromissoryNoteMember
2022-09-12
0001907223
wena:PromissoryNoteMember
2022-09-12
2022-09-12
0001907223
2021-10-10
0001907223
2021-10-10
2021-10-10
0001907223
2022-01-01
2022-12-31
0001907223
2022-11-30
0001907223
2023-01-01
2023-01-31
0001907223
srt:ChiefExecutiveOfficerMember
2024-06-30
0001907223
srt:ChiefExecutiveOfficerMember
2023-12-31
0001907223
us-gaap:SubsequentEventMember
2024-08-12
0001907223
us-gaap:CommonStockMember
2024-06-16
2024-06-21
iso4217:USD
iso4217:USD
xbrli:shares
xbrli:shares
xbrli:pure
iso4217:EUR
ANEW Medical, Inc.
ANEW Medical, Inc., a Delaware
corporation (the “Company”), has filed with the Securities and Exchange Commission (the “SEC”) a Registration
Statement on Form S-1 (the “Registration Statement”) for the purpose of registering for resale under the Securities Act of
1933, as amended (the “Securities Act”), by the selling stockholders named in the prospectus contained in the Registration
Statement an aggregate of 21,527,925 shares including 12,030,000 shares issued upon the exercise of warrants (the “Shares”)
of its common stock, par value $0.0001 per share (the “Common Stock”).
We have examined originals
or copies of such documents, corporate records, certificates of public officials and other instruments as we have deemed necessary or
advisable for the purpose of rendering this opinion.
In rendering the opinion expressed
herein, we have, without independent inquiry or investigation, assumed that (i) all documents submitted to us as originals are authentic
and complete, (ii) all documents submitted to us as copies conform to authentic, complete originals, (iii) all signatures on all documents
that we reviewed are genuine, (iv) all natural persons executing documents had and have the legal capacity to do so, (v) all statements
in certificates of public officials and officers of the Company that we reviewed were and are accurate and (vi) all representations made
by the Company as to matters of fact in the documents that we reviewed were and are accurate.
Based upon the foregoing,
and subject to the additional qualifications set forth below, we advise you that, in our opinion, as of the date hereof, the Shares will
be validly issued, fully paid and non-assessable.
Without limiting any of the
other limitations, exceptions and qualifications stated elsewhere herein, we express no opinion with regard to the applicability or effect
of the laws of any jurisdiction other than the corporate laws of the State of Delaware and the laws of the State of New York, as currently
in effect (based solely upon our review of a standard compilation thereof). This opinion letter deals only with the specified legal issues
expressly addressed herein, and you should not infer any opinion that is not explicitly stated herein from any matter addressed in this
opinion letter.
We hereby consent to the filing
of this opinion as an exhibit to the Registration Statement and further consent to the use of our name therein. In giving this consent,
we do not admit that we are in the category of persons whose consent is required under Section 7 of the Securities Act.
To the Board of Directors and Shareholders of ANEW Medical, Inc.
We hereby consent to the filing of Form S-1, Registration Statement
Dated September 4, 2024.
